
Ja... Ich habe auch nicht die Zeit ewig herum zu synthetisieren, sonst bekomm ich keine Ergebnisse hin. Ich brauche das Keton eben als Edukt für meine Masterarbeit. Leider kosten 500mg stolze 2000 Euro und diesen finanziellen Aufwand will ich umgehen. Wenn man da einen 100 mg Ansatz machen würde und der nicht funktioniert, sind 400 Euro weg. Das ist ja reiner Psychoterror. Ich denke mit der Synthese komme ich vieel günstiger weg, sofern alles einigermaßen klappt.
Bis jetzt wollte ich den Weg über den Ester gehen der von einem DMSO Anion nukleophil angegriffen wird. Das Produkt kann dann mit TFA zum Methylthio-2-Tetralon zyklisiert werden und der Thioether am Schluss mit Pd/C wegreduziert werden (auch wenn ich Bedenken wegen Katalysatorvergiftung habe). Das wären dann vier Schritte, dafür braucht man kein Diazomethan oder so.
Auf der anderen Seite war diese Propionsäure auch nicht sehr günstig und ich frage mich wieviel von diesen 10g am Schluss noch übrig bleiben. Bei dem Naphtolweg könnte man halt wirklich mit 50g oder so anfangen weil das Zeug so verdammt billig ist ... Warscheinlich werde ich am Naphtolweg arbeiten, wenn ich gerade mal Leerlaufzeiten habe ...
Retrosynthese
Moderator: Moderatoren
-
Calciumcitrat
- Illumina-Mitglied
- Beiträge: 170
- Registriert: Montag 11. April 2011, 16:57
-
Calciumcitrat
- Illumina-Mitglied
- Beiträge: 170
- Registriert: Montag 11. April 2011, 16:57
Ah stimmt, denn hatte ich übersehen. Wobei hier die Frage ist, was es bedeutet wenn der Autor die anderen Synthesen als "praktisch überzeugender" beschreibt...
Die Regioselektivität der Bromierung sollte sehr gut sein. Scheint auch die klassische Herangehensweise zu sein: http://orgsyn.org/demo.aspx?prep=CV3P0132
Darf man fragen, in welche Richtung es dann in der Masterarbeit gehen soll? Morphinderivate oder syn. Katecholamine?
Die Regioselektivität der Bromierung sollte sehr gut sein. Scheint auch die klassische Herangehensweise zu sein: http://orgsyn.org/demo.aspx?prep=CV3P0132
Darf man fragen, in welche Richtung es dann in der Masterarbeit gehen soll? Morphinderivate oder syn. Katecholamine?
Hi Leute! Habe nun 22g Dibromnaphthylmethylethers vor mir liegen. Gerade habe ich den dritten Schritt probiert (Methoxylierung) mit einer kleinen Menge. Der Naphthylether ist aber nicht ganz rein, auf der DC (Hex/EtOAc 9:1) zeigen sich ein großer und 2 kleinere Spots. Diese liegen so nahe beieinander, dass ich sie per Säulenchromatographie nicht trennen konnte (was irgendwie auch zu erwarten war). Da aber nach dem dritten Schritt eh gesäult werden muss, überlege ich mir schon die Reinigung nach dem zweiten Schritt zu sparen... NMR-Spektrum hab ich auch aufgenommen, wollt ihr es mal sehen?^^
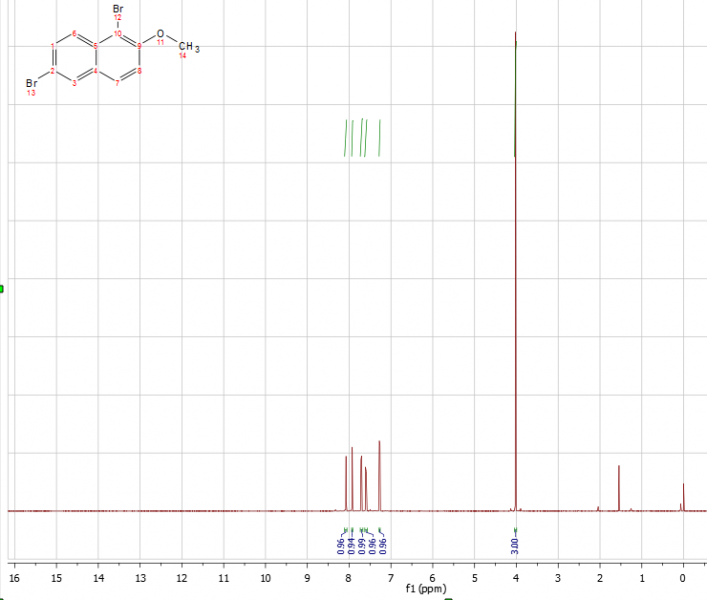
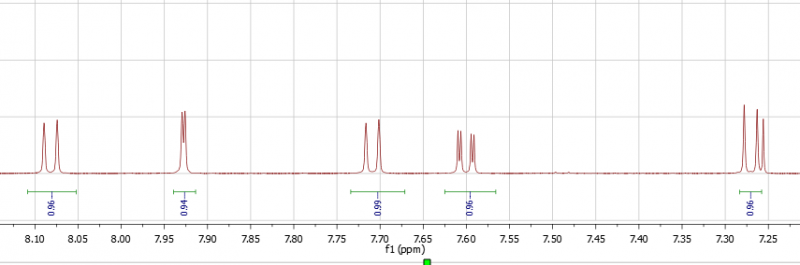
Hinweis: Chloroformpeak bei 7.26 und Wasserpeak bei 1.55 sind nicht integriert.
Was sagt ihr dazu?
PS: Die Synthese mit dem Diazomethan bzw. über das DMSO hab ich erstmal verschoben, weil sie momentan zu langwierig und "nervig" erscheint. Das ist ja alles nur ein Nebenprojekt und gar nicht meine eigentliche Aufgabe Auch wenn wir sonst die Substanz kaufen müssten... Habe bisher nur den Ethylester der Dimethoxypropionsäure synthetisiert.
Auch wenn wir sonst die Substanz kaufen müssten... Habe bisher nur den Ethylester der Dimethoxypropionsäure synthetisiert.
Hinweis: Chloroformpeak bei 7.26 und Wasserpeak bei 1.55 sind nicht integriert.
Was sagt ihr dazu?
PS: Die Synthese mit dem Diazomethan bzw. über das DMSO hab ich erstmal verschoben, weil sie momentan zu langwierig und "nervig" erscheint. Das ist ja alles nur ein Nebenprojekt und gar nicht meine eigentliche Aufgabe
-
Calciumcitrat
- Illumina-Mitglied
- Beiträge: 170
- Registriert: Montag 11. April 2011, 16:57
-
Glaskocher
- Illumina-Mitglied
- Beiträge: 2549
- Registriert: Dienstag 27. Oktober 2015, 22:17
- Wohnort: Leverkusen
Das NMR sieht gut aus. Es sind, bis auf eine Spur Silicon (aus der Spritze?) kaum Verunreinigungen drin. Das Signal der Methoxygruppe ist im erwarteten Bereich und die Zahl/Fläche der Aromaten-H-Signale stimmt auch. Jetzt müßte man nur noch, über die Kopplungskonstanten und eventuell weitere 2D-NMRs diese Signale den einzelnen Positionen/13C-Signalen zuordnen. Die Signale bei 7,93ppm und 7,6ppm scheinen eine 4J-Kopplung (1-2Hz?) zu sein, die drei Anderen und das Signal bei 7,6ppm sind wohl das Ergebnis von 3J-Kopplungen (~7Hz?). Jetzt müßte man nur noch herausfinden, "wer mit wem" und ob es keine Kopplungen über "heterosubstituierte Kohlenstoffe" hinweg gibt, um die Signale eindeutig zuordnen zu können. Ein Vergleich mit Literaturwerten reicht allerdings auch, um die Identität zu bestätigen.
Dann mal gutes Gelingen für die Folgestufen!
Dann mal gutes Gelingen für die Folgestufen!
-
Glaskocher
- Illumina-Mitglied
- Beiträge: 2549
- Registriert: Dienstag 27. Oktober 2015, 22:17
- Wohnort: Leverkusen
Im NMR sieht die Substanz nach mindestens 98% aus. Da würde ich mir um die Spots im DC weniger Sorgen machen, da Naphtenderivate auch sehr farbstark sein können. Falls Du Zugang zu GC oder GC-MS hast, dann miss die Probe auch mal damit. Sie wird die Trennung bestimmt gut überstehen und man kann die Signale von FID- und MS-Detektoren als gute Näherung für Gewichtsprozente nehmen, solange die Substanzen recht ähnlich sind.
-
Calciumcitrat
- Illumina-Mitglied
- Beiträge: 170
- Registriert: Montag 11. April 2011, 16:57
Ups, hab deinen Post oben gar nicht mehr gesehen @Glaskocher. Sind die 4J-Kopplungen zwischen dem H-Atom an C1 und C3 und umgekehrt? Ich bin leider noch nicht so versiert im NMR-Spektren auswerten.
Habe Zugang zu HPLC-MS und könnte am Montag nochmal ein Spektrum messen... wobei ich mich mit 98% sowieso zufrieden geben würde.
Habe Zugang zu HPLC-MS und könnte am Montag nochmal ein Spektrum messen... wobei ich mich mit 98% sowieso zufrieden geben würde.
Danke! Ich hoffe ich bekomme 1000-2000 mg heraus...Dann mal gutes Gelingen für die Folgestufen!
Übrigens: Momentan arbeite ich mit dem anderen Isomer, also dem 6,7-Dimethoxy-2-tetralon, welches auch eher teuer ist. Jetzt habe ich eine relativ simple Synthese entdeckt, die von 3,4-Dimethoxyphenylessigsäure ausgeht: Über das Säurechlorid, das direkt mit Ethengas und Aluminiumchlorid zum Tetralon zyklisiert wird. Klingt ja eigentlich fast zu einfach oder? Was ist davon zu halten? War natürlich schon wieder übermütig und hab das Säurechlorid synthetisiert...

EDIT: Und ja langsam wird es wieder OT...
EDIT: Und ja langsam wird es wieder OT...
-
Glaskocher
- Illumina-Mitglied
- Beiträge: 2549
- Registriert: Dienstag 27. Oktober 2015, 22:17
- Wohnort: Leverkusen
4J-Kopplung: Ich vermute, daß die zwischen den C-Atomen "3" und "7" ist, weil dort kein mit einem "fremden" Atom verbundener Kohlenstoff dazwischen liegt. Die 3J-Kopplungen sind zwischen "1" und "6", beziehungsweise "7" und "8".
Nebenbei: Üblicherweise nummeriert man das Naphthalin-System etwas anders: Beim Brom "12" wird der Kohlenstoff mir der 1 nummeriert, die O-CH3-Gruppe ist an der 2, gegenüber der 1 liegt die 4 und die 5 ist die "untere" CH-Gruppe in zweiten Ring, an der 6 ist das zweite Brom, das erste nummerierte Kohlenstoffatom, das zu beiden Ringen gehört liegt zwischen der 8 und der 1 (mit 9 beziffert) und zwischen der 4 und der 5 dann die 10. Folglich ist das oben (im NMR) gezeichnete Molekül ein 1,6-Dibrom-2-methoxynaphthalin.
Beim Synthesevoeschlag mit dem 3,4-Dimethoxyphenyl-acetylchlorid gehe ich von einer möglichen Umsetzung aus. Du wirst aber mindestens noch ein zweites Isomer in geringerer Menge bekommen, bei dem der zweite Ring zur "anderen Seite" gebildet worden ist. Bei ähnlichen Reaktionen hatte ich mit BCl3 als Kat gearbeitet. Dabei ging es um die Bildung hoch substituierter Indacene.
PS: Zur Not trennen wir diesen Synthesekomplex ab und gönnen ihm einen eigenen Thread. Aber sooo OT ist das doch noch nicht...
Nebenbei: Üblicherweise nummeriert man das Naphthalin-System etwas anders: Beim Brom "12" wird der Kohlenstoff mir der 1 nummeriert, die O-CH3-Gruppe ist an der 2, gegenüber der 1 liegt die 4 und die 5 ist die "untere" CH-Gruppe in zweiten Ring, an der 6 ist das zweite Brom, das erste nummerierte Kohlenstoffatom, das zu beiden Ringen gehört liegt zwischen der 8 und der 1 (mit 9 beziffert) und zwischen der 4 und der 5 dann die 10. Folglich ist das oben (im NMR) gezeichnete Molekül ein 1,6-Dibrom-2-methoxynaphthalin.
Beim Synthesevoeschlag mit dem 3,4-Dimethoxyphenyl-acetylchlorid gehe ich von einer möglichen Umsetzung aus. Du wirst aber mindestens noch ein zweites Isomer in geringerer Menge bekommen, bei dem der zweite Ring zur "anderen Seite" gebildet worden ist. Bei ähnlichen Reaktionen hatte ich mit BCl3 als Kat gearbeitet. Dabei ging es um die Bildung hoch substituierter Indacene.
PS: Zur Not trennen wir diesen Synthesekomplex ab und gönnen ihm einen eigenen Thread. Aber sooo OT ist das doch noch nicht...
Danke dir! MestreNova hat die Nummerierung automatisch gemacht und ich hab mich daran nicht gestört.
Hab eine Testreaktion für die Methoxylierung gemacht und vom aufgearbeiteten, getrockneten Produkt NMR aufgenommen. Resultat: Keine Umsetzung! Naja gut auf der DC hat man auch schon keine Veränderung gesehen, keine Ahnung warum...
Naja gut auf der DC hat man auch schon keine Veränderung gesehen, keine Ahnung warum...
Hab eine Testreaktion für die Methoxylierung gemacht und vom aufgearbeiteten, getrockneten Produkt NMR aufgenommen. Resultat: Keine Umsetzung!