Die hier vorgestellte Synthese lässt leicht und lösemittelfrei die Darstellung zahlreicher Thiocarbonsäureamide aus Carbonylverbindungen zu und stellt den ersten Schritt im Willgerodt-Kindler-Verfahren zur Herstellung von Phenylessigsäuren aus durch Friedel-Crafts-Acylierung leicht zugänglichen Acetophenonen dar. Neben dieser Anwendung sind die Thiosäureamide wichtige Ausgangsstoffe in der Heterocyclensynthese, etwa die Phenylessigsäurederivate zur Herstellung hochsubstituierter Thiophene.
Geräte:
Magnetheizrührer, Silikonölbad, 100 ml Kolben, Rückflusskühler, zwei Gaswaschflaschen (fakultativ), Thermometer, Stativmaterial, Saugflasche und Büchnertrichter etc.
Chemikalien:
Acetophenon
Morpholin
Schwefel
Ethanol
Phenylthioessigsäuremorpholid

Hinweis:
Als Beiprodukte entstehen diverse Organoschwefelverbindungen sowie Schwefelwasserstoff, was zu starker Geruchsbelästigung führt und die Gesundheit schädigen kann. Es ist daher unter einem Abzug zu arbeiten.
Durchführung:
In einen 100 ml Rundkolben werden 6,41 g fein gepulverte Schwefelblüte, 12 g Acetophenon und 12,4 g Morpholin eingewogen und unter Rückflusskühlung im Ölbad auf 135 °C Badtemperatur erhitzt. Die zunächst orange gefärbte Suspension nimmt bereits nach wenigen Minuten eine dunkelrote Farbe an. Allmählich geht dabei der Schwefel in Lösung und das Morpholin beginnt im unteren Teil des Kühlers zu kondensieren (Anm. 1). Nach 6-7 Stunden wird geringfügig abkühlen gelassen und unter Rühren in 40 ml bereits vorgewärmten Ethanol gegossen. Dabei fällt nicht umgesetzter Schwefel aus, welcher zunächst nicht abgetrennt wird. Währen des Abkühlens kristallisiert das Thioamid aus, wobei die Kristallisation durch Kühlung über Nacht vervollständigt wird. Der Kristallbrei wird auf dem Büchnertrichter abfiltriert und mit wenig kaltem Ethanol gewaschen. (Anm. 2)
Nachfolgend wird zwecks einer Umkristallisation der Filterückstand in etwa 50 bis 60 ml siedendem Ethanol aufgenommen, wobei unlösliches Material (Anm. 3) durch heiße Filtration abgetrennt wird. Filtrat und Waschethanol des Rückstands engt man anschließend auf 40-50 ml Volumen ein und lässt langsam abkühlen. Dabei kristallisiert das Produkt in langen Nadeln aus. Nach Filtration wird mit wenig kaltem EtOH nachgewaschen, sodass das Präparat rein weiß gefärbt ist.
Ausbeute: 17,74 g (80,2% d.Th.)
Identität Smp: 78 °C (Lit.: 77-79 °C)
Entsorgung:
Alle Abfälle werden den halogenfreien, organischen Abfällen zugeführt. Kontaminierte Geräte (insb. der Kühler) werden mit alkalischer Permanganatlösung entgiftet. Rückstände im Kolben können leicht mit einer Behandlung im Ultraschallbad beseitigt werden.
Erklärung:
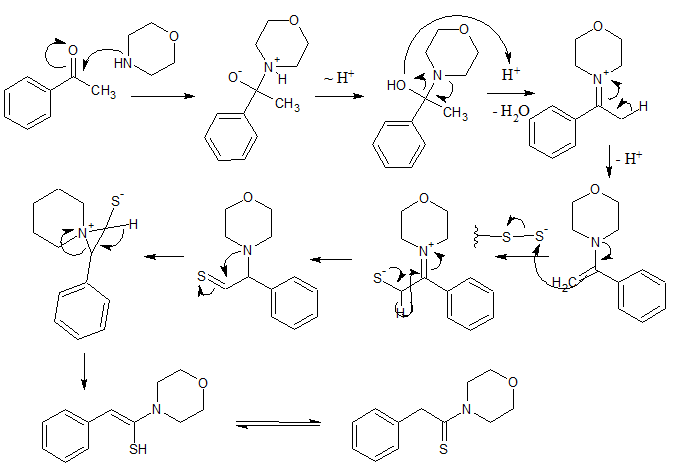
Das Amin greift initial an den Carbonyl-C an und es entsteht ein Enamin. An dessen Doppelbindung addiert Schwefel elektrophil. Der genaue Mechanismus ist - soweit mir bekannt - nicht aufgeklärt, im Mechanismus (unten) wird es als Polysulfid dargestellt (Anm. 4). Aus folgenden Umlagerungen mit Hydrid-Verschiebung geht ein 2-Aminothial hervor, welches intramolekular durch das nicht bindende Elektronenpaar des Stickstoffs nucleophil angegriffen wird. Dieses Aziridimium-thiolat geht unter Ringöffnung und Tautomerisierung in das Thiosäureamid über.
Mechanismus:
Bilder:

Das Setup

Ansatz nach Ablauf der Reaktionszeit

Nach Einguss in Ethanol wird Kristallisation auf dem Schwefelrückstand beobachtet

Am Beginn der Rekristallisation, die Gestalt der Kristalle ist gut erkennbar

Das Präparat
Literatur:
Organikum 22. Auflage, Weinheim 2004, p.427






