Ausgehend von den beiden Vorvorspielen
https://illumina-chemie.org/preparation- ... t3214.html
https://illumina-chemie.org/synthese-nnn ... t3207.html
brachte ich nun diese beiden Edukte zur Reaktion.
Verwendete Vorschrift:
EXAMPLE 2
Preparation of 3,5-Dichloro-2-Hydroxy-N-(2-Dimethylaminoethyl)-N-Methylbenzenesulfonamide
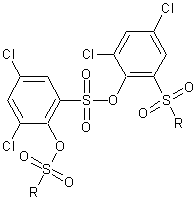
A clarified solution of 3,5-dichloro-2-hydroxybenzenesulfonyl chloride (47.64 grams; 0.18 mole) in 200 mls. of diethyl ether was added to a stirred solution of N,N,N'-trimethylethylenediamine (39.8 grams; 0.39 mole) in 200 mls. of diethyl ether over a period of 105 minutes and the reaction mixture was stirred at room temperature overnight.
The resulting precipitate was separated by filtration, washed with diethyl ether, reslurried in water, separated by filtration, washed with a small amount of water and dried to obtain 53.8 grams (91.5% yield) of crude product which melted at 197° to 200° C. Recrystallization of the crude material from 440 mls. of 50% aqueous ethanol gave 46.1 grams (78.3% yield) of pure material, m.p. 198°-200° C.
http://www.freepatentsonline.com/4053430.html
Da ich nur 17 g Sulfochlorid zur Verfügung habe, war dies der limitierende Faktor, so dass sich folgende Berechnung ergab:
14.20 g des Amins in 71.4 ml Diethylether vorlegen und dazu eine Lösung von 17 g des Sulfochlorids ebenfalls in 71.4 ml Ether gelöst tropfen.....
Hier mal das Sulfochlorid in Ether aufgeschlämmt:

Abwiegen des Amins:

was sich im Ether zu eine klaren Flüssigkeit löst:

Die trübe Sulfochlorid-Lösung wurde sanft (wegen des niedrigen Siedepunkt des Ethers) gefiltert um eine klare Flüssigkeit zu erhalten:

und anschließend durch den Tropftrichter zu der etherischen Aminlösung getropft. Wobei sich die Lösung langsam eintrübte:

Beim Eintropfen zeigte sich an der Eintropfstelle jedes mal eine intensive Gelbfärbung, mit einem Farbton der vergleichbar mit der Farbe von Pikrinsäure ist. Dieselbe Farbe trat auch auf, als Reste des Sulfochlorids beim spülen mit der alkalischen Spüllösung (Deconex Ersatz für Chromschwefelsäure) in Kontakt gekommen sind:


Im weiteren Verlauf schied sich im Kolben eine flüssige Phase ab (leider kein Bild gemacht) die sich unten an sammelte, wobei ich schon die Befürchtung hatte, das da etwas schief gelaufen ist. Allerdings zeigte sich als das gesamte Sulfochlorid in Ether zugetropft worden ist, dass sich eine weiße Substanz abschied wie hier auf dem Bild zu erkennen ist:

Die Reaktion wird über Nacht gerührt und morgen weiter aufgearbeitet.
BJ68