Dibenzalaceton
Geräte:
3-Halsrundkolben, Magnetrührer, Eisbad, Tropftrichter, Thermometer, Rückflusskühler, Messzylinder, Büchnertrichter
Chemikalien:
Benzaldehyd

Ethanol

Aceton
Natriumhydroxid

Dibenzalaceton
Hinweis:
Der erforderliche Benzaldehyd ist zunächst zu reinigen, um die enthaltene Benzoesäure abzutrennen. Dies kann durch Destillation bei Normaldruck oder Vakuumdestillation erfolgen. Frisch gereinigter Benzaldehyd sollte alsbald umgesetzt werden.
Durchführung:
In einen Dreihalsrundkolben mit Tropftrichter und Innenthermometer wird eine Mischung aus 250 mL Wasser und 200 mL Ethanol (96%) vorgelegt. Dann nach werden 25 g Natriumhydroxid eingetragen und gelöst. Ab diesem Schritt muss das Gemisch durch ein Eisbad gekühlt werden (Temp. < 15 °C). Per Tropftrichter gibt man nun ein Gemisch aus 26,5 g/25,4 mL (0,25 mol) frisch destilierten Benzaldehyd und 7,26g/9,25 mL (0,125 mol) Aceton hinzu. Hierbei sollte ca. 1 Tropfen pro Sekunde in den Kolben getropft werden. Es wird mit einem Magnetrührer stark gerührt. Nach 5 Min. wird die Suspension viskoser und trübt sich immer mehr ein. Nach der Zugabe wird noch etwa 20 min. weiter gerührt. Die Innentemperatur sollte bei 10-15 °C gehalten werden.
Der ausgefallene, schwefelgelbe Feststoff wird nun durch Vakuumfiltration abgesaugt und mit kaltem Wasser (Temp. 5 °C) so oft gewaschen, bis der pH-Wert neutral ist. In meinem Fall wurde 3 x mit 15 mL Wasser gewaschen. Dann nach wird noch 1x mit kalten Ethanol (Temp. -10°c) gewaschen. Das Rohprodukt muss nun noch abgesaugt werden. Für ein trockeneres Rohprodukt kann etwas länger gesaugt werden (~5 min).
Da das Produkt noch von Edukten und Nebenprodukten (Benzylalkohol) verunreinigt ist, muss es mindestens 1x umkristallisiert werden. Geeignete Lösungsmittel sind Ethanol, 2-Propanol oder Aceton. Ethylacetat kann auch verwendet werden, dazu muss das Produkt aber zuvor gut getrocknet werden. Das Lösungsmittel wird im Kolben vorgelegt und das Rohprodukt zugegeben. In meinem Fall habe ich Ethylacetat verwendet und habe insgesamt 2x umkristallisiert. Es wird unter Rückfluss sieden gelassen, wobei gerührt wird. Für 20 g Rohprodukt werden ca. 50 mL Ethylacetat benötigt. Sollte nicht das ganze Produkt in Lösung gehen, werden schrittweise weitere 3 mL Ester zugegeben. Die heiße Lösung wird unter Rühren langsam auf Raumtemperatur abgekühlt. Der Ester wird auf einem Büchnertrichter abgesaugt, bis das Produkt trocken aussieht. Letztendlich wird 1x mit kaltem Ethanol (Temp. -10 °c) nachgewaschen (~ 10 mL). Durch Schmelzpunktbestimmung wird die Reinheit bestimmt. Gibt es zu starke Differenzen zwischen selbst gemessenen und Literaturwerten, so muss noch ein weiteres Mal umkristalliesiert werden.
Das Dibenzalaceton wird im Exsikkator über wasserfreiem Calciumchlorid für mehrere Tage getrocknet. Bei mir dauerte es insgesamt 1 Woche. Durch tägliches Wägen des Becherglases samt Inhalt kann bestimmt werden, ob noch Wasser enthalten ist. Ändert sich das Gewicht nicht mehr, so ist es vollkommen getrocknet.
Ausbeute: Rohprodukt: 30 g (107% d.Th.), nach 2 Umkristallisationen: 25,4 g (90% d.Th.)
Schmelzpunktbestimmung: 108-112 °C (Literaturwert: 107-114 °C)
Entsorgung:
Natriumhydroxid wird mit Salzsäure neutralisiert und ins Abwasser gegeben. Ethanol, Aceton, Dibenzylidenaceton und Benzaldehyd kommen in den Behälter "organische Abfälle halogenfrei".
Erklärung:
Diese Reaktion ist eine Kondensationsreaktion, genauer eine Aldolreaktion, durch eine Base katalysiert.
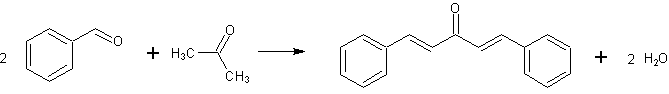
2x 106,12 g/mol + 58,08 g/mol -->234,29 g/mol + 18,01 g/mol
Mein Ansatz:
26,55g g + 7,26 g --> 28,28 g + 2,25 g
Bilder:
(Die Fotos sind von einem anderen Ansatz!)

Die Natriumhydroxid-Lösung

Kühlen des Gemisches

Innentemperatur

Während der Reaktion

Vakuumfiltration
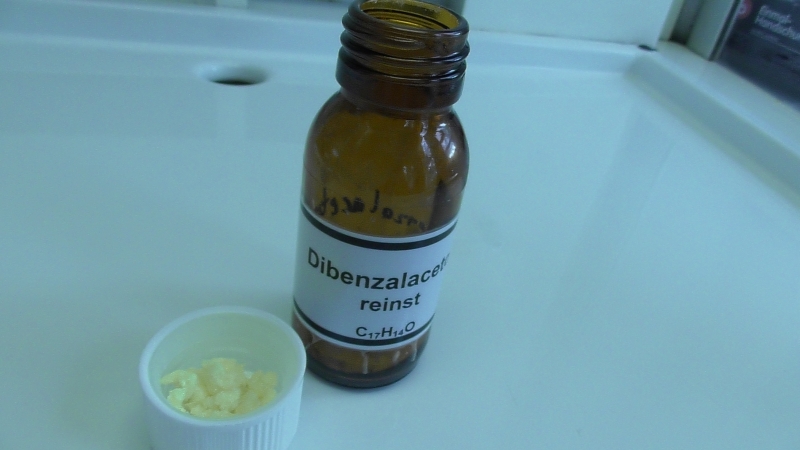
Das Produkt
Literatur:
- Organikum, 22. Auflage

