
Das Übergangsmetall Kobalt bildet mit Ammoniak und weiteren Liganden eine große Zahl von Komplexverbindungen, die im 19. Jahrhundert erstmals dargestellt und systematisch untersucht wurden. Nicht nur die Farbenpracht dieser Salze fesselte die damaligen Chemiker, sie stießen beim Studium ihrer Zusammensetzung auch auf unerwartete theoretische Probleme. Die bunten Komplexsalze wurden zum Ausgangspunkt einer völlig neuen Interpretation der anorganischen Verbindungen und sogar zum Anlass für einen Nobelpreis. Grund genug, sich etwas näher mit ihnen zu beschäftigen.
Drehen wir dazu das Rad der Zeit zurück und machen wir eine Reise in den Winter des Jahres 1892/1893. Wir wollen uns zunächst umsehen, wo die Chemie zu dieser Zeit steht. Wir werden die polytechnische Lehranstalt Kopenhagen und die Universität Zürich besuchen und dort unter fachkundiger Anleitung experimentieren. Zwischen den beiden Epizentren der anorganischen Chemie wird ein Streit um die richtige Deutung der Beobachtungen ausbrechen, die an den “Kobaltammoniakverbindungen“ gemacht werden. Und wir werden der Verleihung des Nobelpreises beiwohnen. Die Zeitmaschine ist mit Chronol, dem Zeit-Treibstoff, betankt (dessen Synthese, nebenbei bemerkt, ungeheuer aufwändig und nicht billig ist!). Es gibt viel zu entdecken und zu experimentieren, weshalb der Reisebericht in mehreren Teilen veröffentlicht wird. Die Reiseleitung empfiehlt: nehmt euch Zeit und macht es euch bequem!
Historische Einleitung:
Im letzten Jahrzehnt des 19. Jahrhunderts hat die Chemie längst die dunklen Laboratorien verlassen, in denen die Alchemisten werkelten. Statt auf dem rußenden Athanor werden die Substanzen über Bunsenbrennern in sauberen Glasgefäßen erhitzt. Praktisch das gesamte moderne Instrumentarium ist vorhanden, wenn auch nicht mit Schliffgeräten (diese werden sich erst in den 30er Jahren des kommenden Jahrhunderts etablieren), sondern mit Kork- und Kautschukverbindungen gearbeitet wird. Die Chemie wird auch nicht mehr, wie noch bis vor wenigen Jahrzehnten, lediglich als Hilfswissenschaft der Medizin und Pharmazie angesehen. Sie ist an allen großen Universitäten und technischen Hochschulen ein eigenständiges und angesehenes Fach.
Vor gut hundert Jahren hatte Lavoisier die Phlogistontheorie endgültig für passé erklärt, die Natur der Verbrennungsvorgänge aufgeklärt und den Chemikern eine Definition des chemischen Elementes an die Hand gegeben, die in den Jahrzehnten nach ihm eine wahre Welle von Neuentdeckungen auslöste. In der zwanziger Jahren erweckte ein englischer Grundschullehrer namens John Dalton die Atomtheorie, die seit Demokrit in einen zweitausendjährigen Dornröschenschlaf verfallen war, zu neuem Leben. Damit konnte er zahlreiche empirische Tatsachen, besonders die stöchiometrischen Gesetzmäßigkeiten der konstanten und multiplen Proportionen, plötzlich auf so einfache Weise erklären, daß seine gewagte Hypothese nach einigen Diskussionen allgemein akzeptiert wurde. Kurz darauf erfindet Berzelius die chemische Formelschreibweise. Seine Formeln haben zwar eine für uns etwas gewöhnungsbedürftige Form, nämlich mit hoch- statt tiefgestellten Indizes, aber ansonsten kommen sie uns schon sehr vertraut vor.
Die weitere Entwicklung der Chemie ist rasant. Praktisch die gesamte klassische Stoffchemie ist ein Kind des 19. Jahrhunderts. Vor kurzem hat Mendelejew die erste Version des periodischen Systems der Elemente vorgestellt. Langsam kommt Ordnung in die Vielzahl der Elemente und Verbindungen. Auch die Analytik hat rasche Fortschritte gemacht und die quantitative Zusammensetzung fast jeder Verbindung kann binnen weniger Tage zuverlässig bestimmt werden. Im Zusammenhang mit der Jagd nach noch unentdeckten Elementen ist die Bestimmung der Atomgewichte von großer Bedeutung. Dazu müssen die Elemente möglichst rein dargestellt werden, was manchmal gar nicht so einfach ist. Komplizierte Trennungsgänge werden ausgearbeitet.
Das Wunderkind des Faches aber ist die organische Chemie. Chemische Fabriken sprießen wie Pilze aus dem Boden. Arzneimittel – besonders Alkaloide und deren halbsynthetische Derivate – und Farbstoffe werden ununterbrochen neu entdeckt, entwickelt und vermarktet. Aspirin und Heroin, synthetischer Indigo, die leuchtenden “Anilinfarben“, der Kunstdünger und nicht zuletzt die Sprengstoffe bescheren der chemischen Industrie einen Aufschwung, der den Vergleich mit der Digitalindustrie unserer Tage nicht zu scheuen braucht.
Großes Thema unter den Fachkollegen ist die chemische Strukturlehre. An den Kohlenstoffverbindungen haben die Chemiker gelernt, dass Isomere existieren - Stoffe gleicher quantitativer Zusammensetzung aber unterschiedlicher Molekülstruktur. Die Strukturaufklärung muss sich im Jahre 1892 – Max von Laue wird die Röntgenstrukturanalyse erst in 20 Jahren entwickeln – noch auf indirekte Schlussfolgerungen aus empirischen Daten stützen. Systematisch werden Substanzen derivatisiert und analysiert um Rückschlüsse auf ihren Aufbau zu ziehen. Bei der Entwicklung der Theorien ist Phantasie gefragt und wird erfolgreich eingesetzt. Alfred Kekulé war die Idee für eine passende Strukturformel des Benzols – eine damals extravagante Ringstruktur – in einem kreativen Halbschlaf, zwischen Träumen und Wachen, zugefallen. Kürzlich hat van’t Hoff "La chimie dans l’espace“ vorgestellt und den Molekülen die dritte Dimension gegeben. Er hat damit nicht nur Zustimmung geerntet, ganz im Gegenteil! Froh, die Wirrungen der spekulativen Naturphilosophie hinter sich gelassen zu haben, melden führende Empiriker wie Hermann Kolbe scharfe Kritik an dem als rein spekulativ gesehenen Entwurf der Tetraederstuktur der Kohlenstoffverbindungen an. Theorien haben sich aus empirischen Tatsachen abzuleiten und sollen nicht am Schreibtisch entwickelt werden! Aber der Ansatz van’t Hoffs erklärt elegant die Existenz der optischen Isomere. Langsam beginnt sich die Vorstellung durchzusetzen, daß Atome und Moleküle nicht nur Konzepte sein sondern womöglich auch physische Realität besitzen könnten.
Die anorganische Chemie nimmt sich neben den grandiosen Erfolgen der Organik etwas blass aus, wenngleich auch auf diesem Gebiet geforscht und publiziert wird, was das Zeug hält. Die Wertigkeit, die Valenz der Atome ist ein wichtiges Strukturprinzip. Fast alle Beobachtungen lassen sich mit der Theorie der Valenz erklären. Auch, dass manche Elemente in mehreren Valenzstufen vorliegen können, ist bekannt. Aber da gibt es einige Substanzen, deren Zusammensetzung und Verhalten mit der herrschenden Strukturtheorie schwierig zu erklären ist.
Dazu gehört das 1822 erstmals von Gmelin dargestellte, orangegelbe Luteokobaltchlorid. Die Analyse ergibt, dass es aus einem Atom Kobalt, sechs Molekülen Ammoniak und drei Chloratomen besteht. Das Kobalt ist dreiwertig. Da gleich gebaute Salze des Chroms, Rhodiums und Iridiums existieren, werden sie allgemein “Luteosalze“ geannt. Die derzeit anerkannte Strukturformel dieser Verbindungen - in der “M“ für ein dreiwertiges Metall steht - sieht so aus.
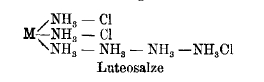
Die uns heutigen unsinnig erscheinende Darstellung fünfbindiger Stickstoffatome stört in unserem Umfeld niemanden. Oktettregel? Die Kollegen schauen die Besucher skeptisch an. Was soll denn das sein? Über den Aufbau des Atoms zu spekulieren entbehrt jeder empirischen Grundlage und auf das Niveau der Naturphilosophie wollen wir uns doch - bitte schön! - nicht begeben! Ist der Stickstoff etwa nicht in vielen Verbindungen fünfwertig, im Ammoniumchlorid beispielsweise, gebunden an fünf einwertige Atome? Und außerdem hat sich das Konzept der Ketten gleicher Atomgruppen in der organischen Chemie bestens bewährt.
Federführend in der anorganischen Chemie sind, in der Nachfolge von Berzelius, die Skandinavier. Christian W. Blomstrand (1826-1897) aus Lund in Schweden hat in seinem Lehrbuch „Chemie der Jetztzeit“ die herrschende Strukturlehre aufgestellt. Jetzt, im Jahre 1892, ist Professor Sophus Mads Jørgensen (1837-1914) in Kopenhagen die führende Autorität auf dem Gebiet der anorganischen Strukturtheorie. Er ist mit Blomstrand befreundet und hat dessen Ansatz weiterentwickelt. Ihn wollen wir zuerst besuchen, denn er verfügt über jahrzehntelange Erfahrung in der Synthese und Analyse der zahlreichen Kobaltsalze, für die wir uns interessieren. Jørgensen eilt der Ruf voraus, ein geschickter Experimentator, ein penibler Analytiker und ein strenger Empirist zu sein. Mit seinem weißen Haar und Bart ist er eine Respekt heischende Erscheinung. Zahllose Schüler, die später ihrerseits berühmte Chemiker werden, hat er ausgebildet.

Schon ein bis zwei Jahrzehnte vor unserem Besuch hatten die Chemiker Erdmann, Gibbs, Rose und andere verschiedene Derivate des Luteokobaltchlorids gefunden, die auf ein Atom des Metalls und drei Atome Chlor nicht sechs, sondern nur fünf oder vier Ammoniakmoleküle enthalten. Da sind zum Beispiel das violettrote Puprpureo- und das grüne Präsokobaltchlorid.

Abb. (v.u.n.o.): Luteokbaltchlorid, Purpureokobaltchlorid und Päsokobaltchlorid
Es war bald aufgefallen, daß diese Salze sich nicht an die bisherigen Regeln der anorganischen Chemie halten. Zum Beispiel kann man durch Silbernitrat aus einer Lösung des Purpureochlorids nur 2/3 , beim Präsokobaltchlorid gar nur 1/3 des enthaltenen Chlors als Silberchlorid ausfällen. Jørgensen hat außerdem gefunden, daß man das Ammoniak in den dreiwertigen Kobaltverbindungen auch durch Ethylendiamin (C2H4(NH2)2) ersetzen kann. Dabei fand er zwei Isomere Ethylendiamin-Kobaltchloride, die grün beziehungsweise violett gefärbt sind.
Wir wollen ihn fragen, wie er das Verhalten dieser Verbindungen erklärt. Aber zuerst sollten wir einige der merkwürdigen Kobaltsalze selbst in Händen halten. Die Anleitungen gibt uns Professor Jørgensen freundlicherweise selbst. In ihrer subtilen Variation der Reaktionsbedingungen und den variablen Ausbeuten erinnern seine Vorschriften an die Synthesen der Organik.
Material/Geräte:
Waage, Messzylinder 10,50 und 100 ml, Bechergläser und Kolben 50-1000 ml, große Abdampfschale, Wasserbad, Ventilator, Magnetrührer, Schliffkolben 100 und 250 ml, Gasentwicklungs-Tropftrichter mit Druckausgleich, Durchbohrter Stopfen mit Gärröhrchen, Kristallisierschale, 2 Waschflaschen 100 und 250 ml, Trichter, Filter, Nutsche mit Absaugvorrichtung, Glasfritte, Uhrgläser, Glasröhren, doppelt durchbohrte Stopfen, Aquarien-Luftpumpe, Schläuche, Reagenzgläser, Tropfpipetten
Chemikalien:
Kobalt(II)-nitrat-4-hydrat



Kobalt(II)-chlorid-6-hydrat
Ammoniaklösung 25 %


Ammoniumchlorid
Ammoniumsulfat
Ammoniumhydrogencarbonat
Kaliumpermanganat
Natriumnitrit
absolutes Ethanol, vergällt

rauchende Salzsäure (37 %)
Salpetersäure 40%
Oxalsäure
Essigsäure
Silbernitratlösung 5 %
konzentrierte Schwefelsäure
Natriumdiphosphat
Kaliumacetat
diverse Kobalt(III)-komplexverbindungen
Sicherheitshinweise:
Kobaltverbindungen stehen im Verdacht, karzinogen zu wirken! Sorgfältig arbeiten, insbesondere das Einatmen von Stäuben vermeiden!
Versuchsdurchführung:
allgemeine Vorbemerkungen:
Bei fast allen folgenden Synthesen muss eine Zeit lang Luft durch eine Lösung geleitet werden. Dazu benutzt man am besten eine kleine elektrische Luftpumpe, z.B. eine Aquarienluftpumpe. Den Luftstrom reguliert man über ein T-Stück, an dessen einem Ausgang ein kurzer Schlauch mit einer Schraubklemme angebracht ist. Als Reaktionsgefäße eignen sich am besten Gaswaschflaschen, für größere Ansätze (Präparate No.2 und 3) verwendet man Kolben mit doppelt durchbohrten Stopfen und entsprechenden Glasrohren, für kleinere (Präparat No. 14) große Reagenzgläser. Die Abluft enthält praktisch immer Ammoniak und wird daher in einen Abzug oder ins Freie abgeleitet.



Abb.: typische Versuchsaufbauten
Bei manchen Synthesen (Präparate No. 12 und 13) muss die Reaktionsmischung “in den Zug gestellt werden“. Damit ist ein Ort gemeint, an dem die Luft frei über die Flüssigkeitsoberfläche hinwegstreichen kann - ein zugiger Platz eben - so dass sie langsam bei niedriger Temperatur eindunstet. Wie man das praktisch realisiert, ist der Kreativität des Experimentators überlassen. Da auch hier aus den Lösungen Ammoniak ausdampft, sind Wohnräume ungünstig, auch sollten die Schalen kindersicher aufgestellt werden. Um eine Verunreinigung zu vermeiden habe ich sie mit einem grobmaschigen Sieb abgedeckt. Den "Zug" habe ich mittels eines Ventilators erzeugt. Natürlich könnte man das Ganze auch in einen gut ziehenden Abzug stellen, den man laufen lassen muss.

Abb: Reaktionsmischung in einer Abdampfschale “in den Zug“ gestellt.
Wenn Lösungen auf dem Wasserbad eingedampft werden müssen (Synthesen No. 2, 3 und 8 ), sollte die Abdampfschale mindestens teilweise in das Wasserbad eintauchen um die Wärmeüberleitung zu verbessern. Muss auf ein bestimmtes Volumen eingedampft werden, so bringt man zweckmäßigerweise vorher mit einem wasserfesten Stift eine Markierung auf der Schale an. Das Abdampfen wird beschleunigt, wenn man mit Hilfe eines Ventilators einen leichten Luftstrom über die Flüssigkeitsoberfläche leitet. Vor allem bei den Präparaten No. 2 und 8 wird dabei Kohlendixoid frei. Das Sprudeln führt zu einer Aerosolbildung und Niederschlag von feinen violetten Tröpfchen auf der "Leeseite", so daß man dort tunlichst eine Abdeckung (Zeitungspapier) auslegt.Am besten führt man das Eindampfen im Freien oder unter einem Abzug durch!

Abb.: Eindampfen auf dem Wasserbad
Für manche Präparate muss ein Chlorid in konzentrierter Schwefelsäure gelöst werden. Die Originalvorschriften geben an, dies in einer Reibschale vorzunehmen.

Abb.: Verreiben von Pentaamminchlorokobalt(III)-chlorid mit konz. Schwefelsäure
Da bei dieser Operation reichlich gasförmiger Chlorwasserstoff frei wird, ist es vorteilhaft, in einem geschlossenen System zu arbeiten, insbesondere wenn zudem noch Salzsäure zugegeben werden muss. Für das Präparat No. 9 habe ich z.B. folgende Apparatur verwendet.

Abb: Zutropfen von konz. Säuren in einer geschlossenen Apparatur
Die Ausgangssubstanz wird mit einem Rührfisch im Kolben vorgelegt, die Säuren durch den Druckausgleichtropftrichter zugegeben und die freiwerdenden Gase über eine Sicherheitswaschflasche, die etwas verdünnte Natronlauge enthält, ins Freie abgeleitet. Wenn man die Mischung anschließend mehrere Tage stehen lassen muss, empfiehlt es sich, den Kolben mit einem Stopfen zu verschließen, der ein mit stark verdünnter Natronlauge gefülltes Gärröhrchen trägt. Eventuell sich noch entwickelnde Gase (HCl, NOx) können so entweichen, werden aber in der Natronlauge absorbiert.

Abb. Kolben mit aufgesetztem Gärröhrchen
Das Auswaschen der erhaltenen Präparate nimmt man am besten auf der Nutsche vor. Die Waschflüssigkeiten variieren von Substanz zu Substanz, die angegebenen Mengen dienen als Richtwerte. Es ist besser, mehrfach mit kleinen Portionen als auf einmal mit einer großen Menge Flüssigkeit zu waschen. Es muss aber vermieden werden, das Präparat zwischen den einzelnen Waschvorgängen ganz trocken zu saugen, damit keine Risse entstehen, durch die nachher das Waschmittel abläuft. Nach der letzten Waschung wird scharf abgesaugt, das Präparat auf einem Uhrglas vom Filterpapier abgeklatscht, mit einem Spatel zerteilt und getrocknet.


Abb.: Absaugen eines Produktes
Manche Präparate werden aus einer (möglichst konzentrierten) wässrigen Lösung durch Zugabe von Ethanol ausgefällt. Hierbei ist es wichtig, das Ethanol langsam und unter sehr gutem Rühren zuzusetzen. Beachtet man dies nicht, kommt es vor, daß sich das Produkt in einer schmierigen Form, vermengt mit Mutterlauge, abscheidet und an den Wandungen und dem Boden der Gefäße haftet. Deshalb sind für Fällungen mit Ethanol immer Bechergläser zu verwenden. Ist das Missgeschick passiert, kann man den Ansatz unter der Mutterlauge über Nacht stehen lassen und auf eine Erstarrung hoffen, worauf das Präparat aus dem Glas gekratzt werden kann. Es muss dann aber zur Reinigung nochmals umgefällt werden. Auch der zum Waschen verwendete Ethanol erzeugt in der Mutterlauge oft - aber nicht immer - einen Niederschlag. Bei manchen Synthesen lohnt es sich, diesen ebenfalls abzusaugen und auszuwaschen. Die so erhaltenen Präparate sind feiner verteilt und daher meist von hellerer Farbe als die aus der Mutterlauge auskristallisierten Substanzen. Sie werden bevorzugt als Ausgangsstoffe für weitere Synthesen verwendet.

Abb.: schmierige Abscheidung von Tetraammincarbonatokobalt(III)-chlorid nach zu schneller Zugabe von Ethanol.

Abb: Tetraammincarbonatokobalt(III)-nitrat, unten aus Mutterlauge kristallisisert, oben mit Ethanol ausgefällt:
Weitere Einzelheiten sind aus den Versuchsbeschreibungen ersichtlich. Anzumerken ist noch, dass dort, wo Lösungen von Ammoniumchlorid und Natriumnitrit hergestellt werden müssen, diese niemals erwärmt werden dürfen! Eine - unter Umständen stürmische - Zersetzung (Stickstoffbildung!) wäre die Folge. Am besten teilt man die zur Lösung angegebene Wassermenge, löst die Salze getrennt auf und vereinigt dann die Lösungen.
Präparate:
1. Hexaamminkobalt(III)-chlorid (Luteokobaltchlorid)
(aus Brauer S. 1532 sowie Jander/Blasius S.339 - s. Literatur)
[Co(NH3)6]Cl3 – Molmasse: 267,4 g/mol
Man verrührt 8 g Kobalt(II)-chlorid-Hexahydrat und 5,4 g Ammoniumchlorid in 8 ml Wasser wobei sich die Salze nicht ganz lösen, fügt dann 18 ml Ammoniaklösung 25 %, worauf eine tiefrote Lösung entsteht, und zuletzt 0,5 g Aktivkohle zu. Dann leitet man für 1½ Stunden einen kräftigen Luftstrom durch die Mischung und fügt währenddessen alle Viertelstunde 2 ml Ammoniaklösung zu. Danach hat sich ein gelber Niederschlag, durchsetzt mit Kohle, abgeschieden und darüber steht eine tiefbraune Flüssigkeit. Man nutscht den Niederschlag ab und verwirft das Filtrat. Der Rückstand wird in 1 %iger, lauwarmer Salzsäure (ca 40°C) gelöst, wozu knapp 100 ml benötigt werden, und über die gleiche Nutsche abgesaugt. Das klare orangegelbe Filtrat wird erwärmt, bis sich ein evtl. ausgefallener Niederschlag löst, dann langsam mit konzentrierter Salzsäure versetzt, bis eine Trübung auftritt (bei meinem Versuch ca. 2-3 ml) und schließlich in einem Eisbad abgekühlt. Es bildet sich glitzernder, feinkristalliner orangefarbener Niederschlag, der sich rasch absetzt. Nach etwa ½ Stunde saugt man ab, wäscht mit 2 x 25 ml Ethanol 60%, dann 3 x 25 ml Ethanol 80%, saugt scharf ab und trocknet bei leicht erhöhter Temperatur (60-80°C).
Ausbeute: 5,2 g (58 %) feinkristallines, orangefarbenes Pulver, leicht wasserlöslich
Im Filtrat fällt durch das zum Waschen verwendete Ethanol weiteres Produkt als gelbes Pulver aus, das wie oben mit wenig Ethanol 60 %, dann mit Ethanol 80% und zuletzt mit absolutem Ethanol gewaschen wird.
Ausbeute: weitere 1,55 g (17,2 %) feines, dunkelgelbes Pulver

Abb: Hexaamminkobalt(III)-chlorid, unten erste Fraktion, oben mit Ethanol gefälltes Präparat
2. Tetraammincarbonatokobalt(III)-nitrat und -chlorid
(nach S.M. Jørgensen; Z. f. anorgan. Chemie 2 (1892): 282-84)
[Co(NH3)4CO3]NO3 + ½ H2O – Molmasse: 257,9 g/mol
[Co(NH3)4CO3]Cl – Molmasse: 222,4 g/mol
Nitrat: Man löst 50 g Ammoniumcarbonat in 200 ml Wasser, gibt zuerst 125 ml Ammoniaklösung 25 % und dann eine Lösung von 25 g Kobalt(II)-nitrat-Tetrahydrat in 50 ml Wasser zu. Durch die tiefviolette Lösung pumpt man für 3 Stunden einen kräftigen Luftstrom, wobei sie sich dunkelrot verfärbt.
Nach Ablauf der Zeit dampft man auf dem Wasserbad auf 150 ml ein, wobei man alle 15 Minuten einen Spatel voll (ca. 3 g) Ammoniumcarbonat zufügt, was jedes Mal ein leichtes Aufbrausen zur Folge hat. Im Ganzen werden so 15 g Ammoniumcarbonat zugesetzt. Dann saugt man ab (auf dem Filter hinterbleibt ein wenig schwarzes Kobalt(II/III)-oxid) und engt das klare, tiefviolette Filtrat auf 100 ml ein, wobei weiter alle 10 Minuten ein Spatel Ammoniumcarbonat zugefügt wird (im Ganzen 5 g). Dann lässt man abkühlen und stellt die Lösung in einer Kristallisierschale in den Kühlschrank. Es kann Übersättigung eintreten. Sollte eine Kristallabscheidung ausbleiben, so rührt und kratzt man mit einem Glasstab an der Gefäßwand. Das abgeschiedene, leuchtend violette Salz wird abgenutscht, auf dem Filter mit etwa 15 ml kaltem Wasser gewaschen und die Mutterlauge mit der Waschflüssigkeit aufgehoben. Dann wird mit 25 ml Ethanol 80% und schließlich mit 3 x 40 ml absolutem Ethanol gewaschen, trockengesaugt und bei Raumtemperatur getrocknet.
Ausbeute: 12,25 g (55,5 %) feinkristallines, leuchtend violettes Pulver; leicht wasserlöslich
Die Mutterlauge wird auf 50-60 ml eingedampft und mit dem 2,5-fachen Volumen absolutem Ethanol versetzt. Es fällt weiteres Produkt als feines, rosaviolettes Pulver aus, das mit wenigen ml Wasser, dann mit Ethanol 80% und zuletzt mit absolutem Ethanol gewaschen wird.
Ausbeute: weitere 2,35 g (10,5 %) feines, dunkelrosafarbenes Pulver
Anmerkung: auf gleichem Wege ist das Tetraammincarbonatkobalt(III)-sulfat zugänglich, wenn man als Ausgangssubstanz statt des Kobalt(II)-nitrates 23,6 g Kobalt(II)-sulfat-Heptahydrat einsetzt.
Zur Synthese des entsprechenden Chlorids verfährt man identisch, geht aber von 20 g Kobalt(II)-chlorid-Hexahydrat aus. Man dampft unter stetem Zusatz von etwa 16 g Ammoniumcarbonat auf 150 ml ein, saugt etwas ausgeschiedenes schwarzes Kobalt(II/III)-oxid ab, dampft weiter bis auf 125 ml ein (nochmals 5 g Ammoniumcarbonat in Portionen zugeben) und verdünnt das Filtrat mit Wasser auf 150 ml. Die abgekühlte Lösung versetzt man erst mit 100 ml Ethanol und fügt dann sehr langsam (25 ml alle 1-2 Minuten) unter gutem Rühren weitere 350 ml Ethanol zu. Die Flüssigkeit trübt sich nach jeder Zugabe von Ethanol rosafarben, wird dann aber wieder dunkelviolett, indem sich das Produkt zusammenballt. Man lässt den Niederschlag einige Stunden unter der Flüssigkeit stehen, saugt dann ab und wäscht wie oben mit sowie 80 %igem und zuletzt absolutem Ethanol und trocknet an der Luft.
Ausbeute: 11,5 g (61,5 %) dunkelviolettes Pulver; leicht wasserlöslich
In der Mutterlauge fällt durch das Ethanol weiteres Produkt als rosafarbener Niederschlag, der sich im Laufe einer Stunde zusammenballt. Er wird ebenfalls abgesaugt und mit absolutem Ethanol gewaschen. Beim Trocknen vertieft sich die Farbe.
Ausbeute: weitere 1,4 g (8 %) feines, rotviolettes Pulver

Abb: Tetraammincarbonatokobalt(III)-nitrat und -chlorid
3. Pentaamminchlorokobalt(III)-chlorid und -hydrogensulfat (Puprpureokobaltsalze)
(nach S. P. L. Sørensen; Z. f. anorgan. Chemie 5 (1894): 369-70; S.M. Jørgensen; J. f. prakt. Chemie [2] 18 (1878): 210-11)
[Co(NH3)5Cl]Cl2 – Molmasse: 250,4 g/mol
[Co(NH3)5Cl]2(HSO4)2SO4 – Molmasse: 648,8 g/mol
Purpureokobaltchlorid: Man löst 20 g Ammoniumcarbonat in einer Mischung aus 100 ml Wasser und 75 ml Ammoniaklösung 25 % und gibt eine Lösung von 15 g Kobalt(II)-chlorid-Hexahydrat in 35 ml Wasser zu. Durch die rotviolette Lösung leitet man für 3 Stunden einen kräftigen Luftstrom, wobei sie sich nach blutrot verfärbt. Man überführt in eine geräumige Abdampfschale, setzt 55 g Ammoniumchlorid zu und dampft auf dem siedenden Wasserbad binnen rund 1 ½ Stunden zu einem Kristallbrei ein. Dabei müssen die sich am Rand absetzenden Krusten mit einem Glasspatel immer wieder gelöst und in die Flüssigkeit gebracht werden.
Den leuchtend violetten Brei lässt man abkühlen, homogenisiert durch Reiben mit einem Pistill und setzt portionsweise (0,5 ml) 25 %ige Salzsäure unter Rühren zu, bis die Mischung schwach sauer reagiert (bei meinem Versuch waren dazu 2 ml Salzsäure nötig. Dabei entweicht unter leichtem Schäumen etwas Kohlendioxid. Die Mischung wird dann mit Wasser in einem Kolben gespült, auf 150 ml verdünnt und mit ca. 0,5 ml Ammoniaklösung 25 % eben alkalisiert (Lackmuspapier!). Man fügt noch 3,0 ml Ammoniaklösung zu und erhitzt den Kolben dann, bedeckt mit einem Uhrglas, für 1 Stunde im siedenden Wasserbad. Danach ist die Flüssigkeit tiefviolett gefärbt und zeigt einen rotvioletten Bodensatz. Anschließend fügt man 100 ml konzentrierte Salzsäure zu und setzt einen Stopfen mit einem Gärröhrchen auf, das mit Wasser und einigen Tropfen Natronlauge beschickt ist. Der Kolben wird für eine weitere halbe Stunde im siedenden Wasserbad erhitzt. Das Gärröhrchen dient nicht nur zur Absorption von Salzsäuredämpfen sondern oft wird auch eine geringe Menge Chlor frei, das im Kolben an der grünlichgelben Farbe erkennbar ist. Die Flüssigkeit färbt sich nun dunkelblau. Nach einer halben Stunde lässt man abkühlen, wobei sich ein reichlicher rosavioletter Niederschlag abscheidet – gelegentlich neben weißen, federartigen Kristallen von Ammoniumchlorid – und die Flüssigkeit sich nahezu entfärbt.

Das Produkt wird abgesaugt, 2-3 x mit 50 ml 12,5 %iger Salzsäure und dann dreimal mit 50 ml absolutem Ethanol gewaschen und trocknen gelassen. Zu erwarten ist eine Ausbeute von 12-13 g. Wenn es deutlich mehr ist (wie es bei meinen Versuchen beidesmal vorkam) enthält das Präparat größere Mengen Ammoniumchlorid. In diesem Fall verrührt man 5 Minuten in 50-60 ml 10 %iger Salzsäure und saugt nach dem Lösen des Ammoniumchlorids erneut ab. Das Filtrat ist fast farblos. Man wäscht wie oben aus und trocknet.
Ausbeute: 12,6 g (79,7 %) feines, leuchtend violettes Pulver; 1 Teil löst sich in 250 Teilen Wasser von 11-12 °C

Abb.: Pentaamminchlorokobalt(III)-chlorid
Das Präparat kann als Verunreinigung kleine Mengen Hexaamminkobalt(III)-chlorid enthalten. Wenn man eine Probe mit warmem Wasser verrührt und das Filtrat mit konzentrierter Natriumdiphosphatlösung versetzt, darf kein Niederschlag auftreten (Hexaamminkobalt(III)-diphosphat).
Zur Darstellung des Sulfates verreibt man in einer Reibschale 2,5 g Pentaamminchlorokobalt(III)-chlorid mit 3,5 ml konzentrierter Schwefelsäure. Unter Schäumen entweicht Chlorwasserstoff (Abzug!) und es entsteht eine rotviolette Paste, die man 10 Minuten stehen lässt, bis der HCl weitgehend ausgegast ist. Dann löst man in 20 ml heißem Wasser (ca. 70 °C) und stellt die dunkelviolette Lösung in einem kleinen Bechergläschen für mehrere Tage beiseite. Während dieser Zeit werden die zunächst ausgefallenen feinen Kristalle größer. Man saugt auf der Fritte ab und wäscht das Präparat mit 4-5 x 15 ml absolutem Ethanol nach. In der Mutterlauge fällt durch das Ethanol eine weitere Produktfraktion als rosarotes Pulver aus, die ebenfalls abgesaugt und gewaschen wird.
Ausbeute: 1,62 g (50 %) glänzende, dunkelrote Kristallnadeln sowie 1,35 g (40%) feines rosafarbenes Pulver

Abb.: Pentaamminchlorokobalt(III)-hydrogensulfat
Löst man eine Reagenzglasrundung des Präparates in 5 ml Wasser und etwas Schwefelsäure und gibt Silbernitralösung zu, so tritt zunächst keine Fällung ein. Erst beim Erhitzen der Mischung bildet sich ein allmählich dichter werdender Niederschlag von Silberchlorid.

Abb: Lösung von Pentaamminchlorokobalt(III)-sulfat mit Silbernitrat links frisch hergestellt, rechts nach kurzem Erhitzen
4. Pentaamminaquokobalt(III)-oxalat und -sulfat (Roseokobaltoxalat bzw. -sulfat)
(nach S.M. Jörgensen; Z. f. anorgan. Chemie 17(1898): 460-462)
[Co(NH3)5H2O]2(C2O4)3 – Molmasse: 587,8 g/mol
[Co(NH3)5H2O]2(SO4)3 – Molmasse: 611,8 g/mol
Man übergießt in einem 100 ml-Erlenmeyerkolben 2,5 g Pentaamminchlorokobalt(III)-chlorid mit einer Mischung aus 26 ml Wasser und 4 ml Ammonniaklösung 25 % und erhitzt im Dampfraum eines siedenden Wasserbades (nicht im Wasserbad) unter gutem Rühren so lange, bis eine klare, tiefviolette Lösung entstanden ist. Man saugt durch eine Nutsche von möglichen Verunreinigungen (Kobaltoxid) ab und lässt das Filtrat abkühlen. Dann setzt man unter Rühren portionsweise (je 5 ml) 10 %ige Oxalsäurelösung zu, bis sich die Farbe nach Rot aufhellt und ein Niederschlag auszufallen beginnt (bei meinem Versuch 20 ml Oxalsäurelösung). Nun fügt man langsam (ml-weise) weiter Oxalsäure zu, bis eine Tüpfelprobe auf Lackmuspapier eben saure Reaktion anzeigt, lässt eine halbe Stunde stehen, saugt ab und wäscht das Produkt mit 4 Portionen zu je 40-50 ml kaltem Wasser nach. Es wird auf der Heizung getrocknet.
Ausbeute: 2,7 g (82,3 %) feines, hellrotes Pulver, schwer wasserlöslich

Abb.: Pentaamminaquokobalt(III)-oxalat
Zur Darstellung des Sulfates verrührt man 5 g des Oxalates in 20 ml Wasser und fügt 30 ml Schwefelsäure 25% hinzu, worauf eine klare, dunkelrote Lösung entsteht. Diese versetzt man mit dem gleichen Volumen Ethanol und saugt den ausfallenden Niederschlag nach kurzem Stehen ab. Man wäscht 3-4 x mit 25 ml Ethanol 50 % und danach mit 2 x 15 ml absolutem Ethanol aus und trocknet an der Luft
Ausbeute: 4,9 g (95%) feines rosastichig-orangerotes Pulver, wenig wasserlöslich.
5. Pentaamminaquokobalt(III)-chlorid (Roseokobaltchlorid)
(aus Biltz/Biltz S. 168, s.Literatur)
[Co(NH3)5H2O]Cl3 – Molmasse: 268,4 g/mol
10 g Kobalt(II)-chlorid-Hexahydrat werden in 180 ml Wasser gelöst und 55 ml Ammoniaklösung 25 % zugegeben. Nach Zugabe einer Lösung von 5 g Kaliumpermanganat in 200 ml Wasser lässt man verschlossen 24 Stunden stehen, während derer die Mischung ab und zu umgeschüttelt wird. Dann gießt man die überstehende, rotviolette Flüssigkeit soweit wie möglich von dem braunen Mangandioxidschlamm ab und saugt mehrfach durch dasselbe Filter ab. Der Niederschlag wird ebenfalls abgesaugt (langwierig!), die Filtrate vereinigt und für 6 Stunden in den Kühlschrank gestellt. Danach gibt man in kleinen Portionen (5 ml) verdünnte Salzsäure (25 %) zu, bis eben saure Reaktion eintritt (bei meinem Versuch waren dazu zwischen 35 und 40 ml notwendig) und kühlt in einem Eis-Kochsalz-Bad stark ab. Nun gibt man portionsweise eine – ebenfalls gekühlte – Mischung aus 375 ml konzentrierter Salzsäure und 125 ml Ethanol zu, wobei die Temperatur möglichst nicht über 10 °C steigen sollte. Es fällt ein hochroter Niederschlag aus, den man nach ¼ Stunde absaugt und in Portionen mit ca. 250 ml absolutem Ethanol nachwäscht. Tockensaugen und bei Zimmertemperatur trocknen.
Ausbeute: 3,6 g (32 %), zinnoberrotes, feines Kristallpulver.
Im Tiefkühlfach aufbewahren, da bei Raumtemperatur im Laufe der Zeit, unter Verfärbung nach Violett, eine Umwandlung in Pentaamminchlorokobalt(III)-chlorid eintritt!


Abb: Pentaamminaquokobalt(III)-chlorid
oberes Bild: kurz nach Darstellung, das große RG wurde im Gefrierfach aufbewahrt, das kleine bei Raumtemperatur - unteres Bild: Präparate 8 Wochen alt
Wie man auf dem nächsten Bild sieht hatten schon frühere Chemikergenerationen mit diesem Problem zu tun:

Abb.: historisches Präparat aus den 1970er Jahren: ehemals Pentaamminaquokobaltchlorid, später umbeschriftet in Pentaamminchlorokobaltchlorid!
Anmerkung: die nach diesen Vorschriften dargestellten Pentaamminaquokobaltsalze enthalten oft kleine Mengen Hexaamminkobalt(III)-salze als Verunreinigung. Das Oxalat ist davon leicht zu reinigen, indem man es in 2 %igem Ammoniakwasser löst, wobei Hexaamminkobalt(III)-oxalat ungelöst bleibt, filtriert und das Filtrat erneut mit Oxalsäure fällt. Beim Fällen mit konzentrierter Salzsäure und Ethanol (unter guter Kühlung!) erhält man das reine Chlorid.
6. Pentaamminnitrokobalt(III)-chlorid (Xanthokobaltchlorid)
(nach S.M. Jörgensen; Z. f. anorgan. Chemie 17(1898): 463-64)
[Co(NH3)5NO2]Cl2 – Molmasse: 260,9 g/mol
Man erwärmt 5 g Pentaamminchlorokobalt(III)-chlorid mit 55 ml Wasser und 5 ml Ammoniaklösung 25% im Dampfraum des Wasserbades (siehe Präparat No. 4) bis eine klare, tief rotviolette Lösung entstanden ist und filtriert heiß. Das Filtrat wird in kaltem Wasser abgekühlt und in Portionen von 0,5 ml mit verdünnter Salzsäure (25 %) versetzt, bis die Mischung gegen Lackmus eben sauer reagiert (bei mir waren dazu 6 ml notwendig). Dann fügt man portionsweise 6,25 g Natriumnitrit zu (leichte Entwicklung von NOx – Abzug!), wobei sich die Mischung trübt und die Farbe nach gelbbraun wechselt, und erhitzt, mit einem Uhrglas bedeckt, im siedenden Wasserbad, bis nach 20-30 Minuten eine klare dunkelbraune Lösung erhalten wird. Diese kühlt man in kaltem Wasser ab, wobei eine Kristallabscheidung eintritt, und fügt in Portionen 62,5 ml konzentrierte Salzsäure zu (auch hier werden anfangs Stickoxide frei!). Man stellt den Kolben für eine Stunde in Eiswasser, saugt dann ab, wäscht den Niederschlag mit 2 x 20 ml Salzsäure 20 % und 4 x 25 ml absolutem Ethanol aus und trocknet an der Luft.
Ausbeute: 3,8 g (73 %), ockerbraunes, glitzerndes Kristallpulver; 1 Teil löst sich in 39,2 Teilen Wasser von 21,1 °C bzw in 40,2 Teilen Wasser von 14 °C

Abb.: Pentaamminnitrokobalt(III)-chlorid
7. Tetraammindiaquokobalt(III)-chlorid und -sulfat (Tetraamminroseokobaltsalze)
(nach S.M. Jörgensen; Z. f. anorgan. Chemie 2 (1892): 294)
[Co(NH3)4(H2O)2]Cl3 – Molmasse: 269,4 g/mol
[Co(NH3)4(H2O)2]2(SO4 )2 + 3 H2O – Molmasse: 667,8 g/mol
Chlorid: In einem Becherglas löst man unter Eiskühlung 2 g Tetraammincarbonatokobalt(III)-chlorid in 10 ml Wasser und fügt dann portionsweise 4 ml 25 %ige Salzsäure zu. Die Mischung entwickelt unter Schäumen Kohlendioxid. Nachdem die Gasentwicklung aufgehört hat gibt man, weiter unter guter Kühlung, 4 x 10 ml rauchende Salzsäure zu, wobei man immer 2-3 Minuten stehen lässt. Es fällt ein dunkelroter Niederschlag aus. Nach einer weiteren halben Stunde im Eisbad wird der Niederschlag abgesaugt, mit 4 x 15 ml 95%igem Ethanol gewaschen und getrocknet.
Ausbeute: 1,8 g (74,3 %) dunkelrotes, etwas violettstichiges Pulver; leicht wasserlöslich
Als Ausgangsmaterial kann auch das Tetraammincarbonatokobalt(III)-nitrat und zur Fällung 32 %ige Salzsäure verwendet werden (billiger als rauchende Salzsäure). Man löst 2 g in 8 ml Wasser und gibt 2,5 ml Salzsäure 32 % zu. Nach Ende der Kohlendioxidentwicklung wird wie oben unter guter Kühlung mit 40 ml 32 %iger Salzsäure gefällt. Waschen mit 15 ml Salzsäure, denn mit Ethanol. Das Präparat ist frei von Nitrat. Ausbeute 1,65 g (84%).
Dieses Präparat ist noch unbeständiger als das Pentaamminaquokobalt(III)-chlorid. Bei Zimmertemperatur wechselt die Farbe binnen weniger Tage zu violett, und nach 2 Wochen ist die Substanz vollständig in Tetraamminaquochlorokobalt(III)-chlorid umgewandelt. Auch die hellrote Lösung des Salzes färbt sich beim Stehenlassen allmählich, beim Erwärmen sehr schnell, violett


Abb: Tetraammmindiaquokobalt(III)-chlorid
Oben: frisch dargestellt (kleines RG) - unten: 14 Tage bei Zimmertemperatur gelagert (großes RG)


Abb.: Lösung von Tetraammindiaquokobalt(III)-chlorid (linkes RG) und von Tetraamminaquochlorokobalt(III)-chlorid (rechtes RG) oben frisch bereitet, unten nach kurzem Erhitzen
Zur Darstellung des besser haltbaren Sulfates löst man unter Kühlung 2,4 g Tetraammincarbonatokobalt(III)-nitrat in einer Mischung aus 40 ml Wasser und 4,5 ml verdünnter Schwefelsäure (25 %ig), filtriert und gibt dann in Portionen 25 ml Ethanol zu. Der ausfallende Niederschlag wird abgesaugt und mit 15 ml einer Mischung aus gleichen Volumenteilen Ethanol und 25 %iger Schwefelsäure einige Stunden stehen gelassen. Dann wird erneut abgesaugt, mit 3-4 x 15 ml absolutem Ethanol gewaschen und getrocknet.
Ausbeute: 2,48 g (80 %) rosarotes Pulver; leicht wasserlöslich
Auf Abwesenheit von Nitrat prüft man, indem man eine kleine Menge des Präparates im RG mit 2 ml Wasser und einigen Tropfen Natronlauge kocht, das ausgefalle, schwarze Kobaltoxid abfiltriert und mit dem farblosen Filtrat nach Ansäuern mit Schwefelsäure die Ringprobe durchführt (mindestens ¼ Stunde warten!).
8. Tetraamminaquochlorokobalt(III)-sulfat und –chlorid (Tetraamminchlorokobaltsalze)
(nach S.M. Jørgensen; J. f. prakt. Chemie [2] 42 (1890): 211-13)
[Co(NH3)4H2OCl]Cl2 – Molmasse: 251,4 g/mol
[Co(NH3)4H2OCl]SO4 – Molmasse: 276,4 g/mol
Man löst 50 g Ammoniumcarbonat in 200 ml Wasser, gibt 125 ml Ammoniaklösung 25% zu und gießt unter gutem Schwenken eine Lösung von 20 g Kobalt(II)-chlorid-Hexahydrat in 50 ml Wasser hinein. Durch die Lösung wird dann 2 Stunden lang Luft geleitet. Anschließend wird wie oben (Präparate 2 und 3) unter Zugabe von Ammoniumcarbonat (alle 15 Minuten 1 großer Spatel, im Ganzen ca. 18 g) auf dem Wasserbad auf 100 ml eingeengt, was rund 1 ¾ Stunden in Anspruch nimmt. Die heiße Lösung wird filtriert und abkühlen gelassen. Anschließend setzt man, anfangs in kleinen Portionen, weil die Lösung unter Aufschäumen Kohlendioxid entwickelt, 125 ml Salzsäure 25 % und danach 75 ml rauchende Salzsäure zu. Hierbei kann bereits ein violetter Niederschlag ausfallen. Nun erhitzt man unter ständigem Rühren in einem Becherglas auf dem Dreifuß für etwa 2-3 Minuten, bis die Lösung tiefviolett geworden ist und fast kein Kohlendioxid mehr ausperlt (nicht bis zum Sieden!). Man lässt über Nacht stehen und stellt das Gefäß dann für einige Stunden in den Kühlschrank. Das ausgefallene, rohe Tetraamminaquochlorokobalt(III)-chlorid wird abgenutscht, mit 3 x 20 ml 25 %iger Salzsäure und 4 x 40 ml absolutem Ethanol gewaschen und getrocknet.
Ausbeute: 15,1 g (71,5 %) feines, tiefviolettes Pulver; teilweise wasserlöslich

Abb.: rohes Tetraamminaquochlorokobalt(III)-chlorid
Das Präparat ist nicht rein, sondern enthält Pentaamminchlorokobaltchlorid (Purpureochlorid) sowie etwas Tetraammindichlorokobalt(III) chlorid (Präsochlorid). Letzteres kann man identifizieren, wenn man eine Prise des Kristallpulvers, in Ethanol aufgeschwemmt, unter dem Mikroskop betrachtet. Zwischen den kleinen regulären Oktaedern des Aquochlorosalzes findet man einzelne größere, grüne Kristalle des Dichlorosalzes.

Um das Salz zu reinigen, setzt man es zum Sulfat um. Dazu verrührt man 15 g des Rohproduktes gut mit 450 ml Wasser von 25 °C, dem 1 ml Schwefelsäure (25 %) zugesetzt wurden, und saugt vom Ungelösten ab. Das dunkelviolette Filtrat, dessen Farbe der einer Kaliumpermanganatlösung ähnelt, wird mit einer Lösung von 25 g Ammoniumsulfat in 75 ml Wasser versetzt und 24 Stunden an einem kühlen Ort stehen gelassen. Die ausgeschiedenen Kristalle werden abgesaugt und mit wenig eiskaltem Wasser (3 x 15 ml) gewaschen. Das Waschwasser läuft fast ungefärbt ab.
Ausbeute: 5,15 g (31 %), schwarzviolette Kristalle, schwer wasserlöslich

Abb.: Tetraamminaquochlorokobalt(III)-sulfat
Aus dem Sulfat wird reines Tetraamminaquochlorokobalt(III)-chlorid folgendermaßen erhalten: Man verreibt 3,0 g des Sulfats in einer Reibschale fein, übergießt in einem Kolben mit 30 ml Salzsäure von 25 % und lässt unter gelegentlichem Rühren 24 Stunden bei Raumtemperatur stehen. Man gießt den fast farblosen Überstand vom Bodensatz ab, überführt diesen auf eine Nutsche, wäscht mit 3 x 10 ml Salzsäure von 25% sowie 4 x 10 ml Ethanol nach und trocknet an der Luft. Das Produkt verreibt man erneut, und lässt abermals mit 25 ml Salzsäure 25 % über Nacht stehen (gelegentlich rühren). Nach dem Waschen und Trocknen erhält man ein schwefelsäurefreies Präparat, das mit Bariumchlorid keine Trübung mehr gibt.
Ausbeute: 2,5 g (91,6 %) rotviolettes Kristallpulver; 1 Teil löst sich in 40 Teilen Wasser
Reines Tetraamminaquochlorokobalt(III)-chlorid erhält man auch durch Lagern von Tetraammindiaquokobalt(III)-chlorid (Präparat No. 7) bei Zimmertemperatur für ca. 2 Wochen und anschließendes Trocknen an der Luft.
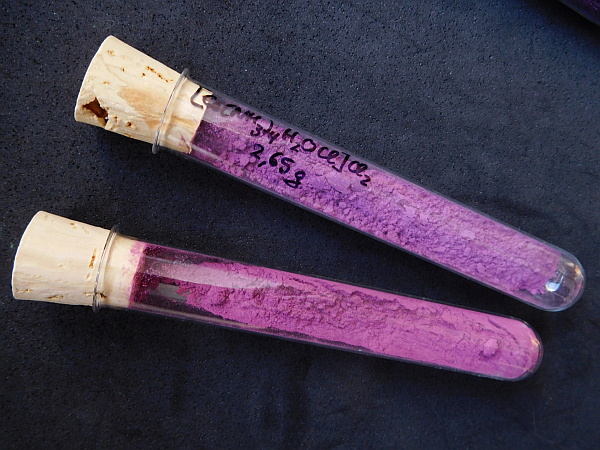
Abb.: Tetraamminaquochlorokobalt(III)-chlorid, oben: durch Lagerung des Diaquokomplexes erhalten, unten: aus dem Sulfat dargestellt
9. trans-Tetraammindichlorokobalt(III)-hydrogensulfat und -chlorid (Präsokobaltsalze)
(nach A. Klein und A. Werner; Z. f. anorgan. Chemie 14(1897): 29; 34 / S.M. Jörgensen; Z. f. anorgan. Chemie 14(1897): 415-17)
trans-[Co(NH3)4Cl2]HSO4 – Molmasse: 294,9 g/mol
trans-[Co(NH3)4Cl2]Cl + H2O– Molmasse: 251,4 g/mol
Zur Darstellung des “sauren Präsokobaltsulfates“ versetzt man in einem Kolben (siehe unter Vorbemerkungen!) 5 g Tetraammincarbonatokobalt(III)-nitrat (oder 4,5 g des Chlorids) unter Rühren allmählich mit so viel konzentrierter Salzsäure, bis kein Kohlendioxid mehr entweicht (etwa 4-4,5 ml). Daraufhin stellt man den Kolben in ein Eisbad und tropft zu dem rosaroten Brei 25 ml konzentrierte Schwefelsäure. Man rührt, bis sich alles gelöst hat und die Chlorwasserstoffentwicklung beendet ist (wenn man das Nitrat verwendet, werden dabei auch Nitrose Gase und etwas Chlor frei, was aber nicht stört). Nun tropft man langsam (binnen 10-15 Minuten) 25 ml konzentrierte Salzsäure zu. Auch dabei tritt anfangs ein kräftiges Aufbrausen ein. Nach kurzer Wartezeit gießt man die blauviolette, trübe Mischung in einen Erlenmeyerkolben, verschließt mit einem Stopfen mit Gärrohr und lässt ruhig stehen. Bereits nach einigen Stunden bildet sich ein grasgrüner Niederschlag aus, der in den nächsten Tagen zunimmt, während der violette Überstand immer transparenter wird.




Abb.: Farbänderung des Reaktionsansatzes
Nach 72 Stunden gießt man den nur noch gering gefärbten Überstand soweit wie möglich ab und spült das Produkt mit etwas 25 %iger Schwefelsäure auf eine Fritte. Dann wird abgesaugt, dreimal mit 10 ml verdünnter Schwefelsäure und 3-4 x mit 15 ml absolutem Ethanol gewaschen und getrocknet.
Ausbeute: 5,4 g (91,6 %) feines, leuchtend grünes Kristallpulver; gut wasserlöslich
Alternativ kann nach Jörgensen auch Tetraamminaquochlorokobalt(III)-sulfat oder –chlorid in den gleichen Mengenverhältnissen verwendet werden. Die Ausbeute ist auch hier sehr gut, wie in einem orientierenden Reagenzglasversuch festgestellt werden konnte..
Die Lösung des Salzes in Wasser ist anfänglich tiefgrün gefärbt. Nach einigen Minuten wechselt die Farbe zu graublau und schließlich zu violett. Gibt man zu der violetten Lösung etwas Ammoniumsulfat, so scheidet sich bei Stehenlassen dunkelviolettes Tetraamminaquochlorokobalt(III)-sulfat aus.

Abb.: Farbwechsel einer Lösung von trans-Tetraammindichlorokobalt(III)-hydrogensulfat
Zur Darstellung des Chlorids (Präsokobaltchlorid) lässt man 4 g des Hydrogensulfates unter gelegentlichem Rühren mit 40 ml 32%iger Salzsäure für 2-3 Tage stehen, saugt ab und verrührt den Rückstand erneut mit Salzsäure. Nach erneuten mehrtägigem Stehen gießt man die blassvioletten Uberstand ab, spült mit etwas Alzsäure auf eine Fritte , saugt ab und wäscht mit 3 x 10 ml konzentrierter Salzsäure und 3-4 x 15 ml Ethanol und trocknet an der Luft.
Ausbeute: 3,2 g (89 %) hellgrünes Pulver, mäßig gut wasserlöslich

Abb.: trans-Tetraammindichlorokobalt(III)-hydrogensulfat und -chlorid
Das Tetraammindichlorokobalt(III)chlorid löst sich in Wasser viel schwerer als das Hydrogensulfat und die Lösung ist wesentlich unbeständiger. In wenigen Sekunden schlägt die Farbe der zunächst grünen Flüssigkeit nach Blaugrau, dann nach Violett und nach einigem Warten nach Rosarot um.
10. cis-Tetraammindinitrokobalt(III)-nitrat und –chlorid (Flavokobaltsalze)
(nach S.M. Jörgensen; Z. f. anorgan. Chemie 5 (1894):162-65 und 17(1898): 472-74)
cis-[Co(NH3)4(NO2)2]NO3 – Molmasse: 280,9 g/mol
cis-[Co(NH3)4(NO2)2]Cl – Molmasse: 254,4 g/mol
Man mischt 70 ml Wasser mit 10 ml Salpetersäure von 40 % (Dichte 1,24 g/ml) und löst darin portionsweise (Vorsicht! schäumt!) unter Umrühren 7,5 g Tetraammincarbonatokobalt(III)-nitrat, ohne zu Erwärmen. Unter Entweichen von Kohlendioxid entsteht eine tief violettrote Lösung, die man unter Rühren in Anteilen mit insgesamt 15 g festem Natriumnitrit versetzt. Dabei schlägt die Farbe unter Abkühlung und Entweichen nitroser Gase (Abzug!) nach Braunrot um. Nun stellt man den Kolben in ein siedendes Wasserbad, bis nach 6-7 Minuten die Gasentwicklung zu Ende gekommen ist und die Mischung eine dunkel-braungelbe Farbe angenommen hat. Man kühlt sofort in kaltem Wasser ab und stellt in Eiswasser. Nun werden unter gutem Rühren portionsweise insgesamt 100 ml Salpetersäure 40 % zugegeben, wobei anfangs erneut nitrose Gase entweichen (anfangs langsam zugeben, kann schäumen!). Die resultierende, braune Lösung, in der schon ein braungelber Niederschlag ausfällt, stellt man über Nacht in den Kühlschrank, saugt die abgeschiedenen Kristalle ab und wäscht zweimal mit 15 ml derselben Salpetersäure nach. Danach wird mit 4 x 30 ml Ethanol 95 % gewaschen und an der Luft getrocknet.
Ausbeute: 6,1 g (74,6 %) curryfarbenes, feines Kristallpulver Kristalle; 1 Teil löst sich in 33 Teilen kaltem Wasser
Zur Darstellung des Chlorids löst man 2,5 g cis-Tetraamminndinitrokobalt(III)-nitrat in 75 ml Wasser, wobei man den Kolben durch kurzes (!) Eintauchen in ein heißes Wasserbad allenfalls leicht erwärmt, gibt 5 g Ammoniumchlorid zu und kühlt in kaltem Wasser ab. Die ganz abgekühlte, klare Lösung versetzt man langsam unter Rühren mit insgesamt 240 ml absolutem Ethanol. Nachdem etwa 2/3 des Ethanols zugegeben worden sind tritt eine gelbe Fällung auf, die sich bald zu einem gelborangenem Niederschlag verdichtet. Nach Stehen im Kühlschrank über Nacht saugt man ab, wäscht mit 4 x 30 ml 95%igem Ethanol nach und trocknet.
Ausbeute: 1,7 g (75,2 %) feines, gelborangefarbenes Kristallpulver; sehr leicht wasserlöslich

Abb.: cis-Tetraammindinitrokobalt(III)-nitrat und - chlorid
11. trans-Tetraamminndinitrokobalt(III)-chlorid (Croceokobaltchlorid)
(nach S.M. Jörgensen; Z. f. anorgan. Chemie 17(1898): 468-70)
trans-[Co(NH3)4(NO2)2]Cl – Molmasse: 254,4 g/mol
In 80 ml Wasser löst man kalt 10 g Ammoniumchlorid und 13,5 g Natriumnitrit, fügt 12 ml Ammoniaklösung 25% und schließlich eine Lösung von 9 g Kobalt(II)-chlorid-Hexahydrat in 20 ml Wasser zu. Durch die dunkelrote Flüssigkeit wird 4 Stunden lang Luft geleitet, wobei sie sich zunächst braun färbt und dann ein braungelber Niederschlag ausfällt. Man lässt 12 Stunden stehen und saugt dann ab. Der Niederschlag wird auf dem Filter zunächst mit 4-5 Portionen (je 25 ml) kaltem Wasser gewaschen, bis eine Probe des ablaufenden Filtrats nach Zusatz von Ammoniumoxalatlösung auch nach mehreren Minuten keinen Niederschlag mehr gibt (Auswaschen von Pentaamminnitrokobalt(III)-salzen). Dazu waren bei mir 4 Waschungen nötig. Anschließend wäscht man 3 x mit 25 ml absolutem Ethanol nach und trocknet an der Luft.
Das Rohprodukt besteht aus einer Mischung des Nitrats und des Chlorids. Erhalten werden 6 g. Es kann direkt zur Darstellung von Tetraamminchloronitrokobalt(III)-chlorid (Präparat No. 18, siehe dritte Zeitreise!) verwendet werden.
Zur Darstellung des reinen Chlorids löst man 3 g des Rohproduktes in einer Mischung aus 60 ml Wasser und 0,2 ml Essigsäure unter Erhitzen (fast bis zum Sieden), filtriert schnell durch einen angewärmten Trichter in einen heiß ausgespülten Kolben und löst im Filtrat 6 g Ammoniumchlorid. Dann wird rasch in Eiswasser abgekühlt, über Nacht im Kühlschrank stehen gelassen und schließlich das ausgefallene Salz abgesaugt. Es wird dreimal mit 25 ml 90 %igem Ethanol nachgewaschen, bis das Waschwasser durch salpetersaure Silbernitratlösung nur noch opaleszierend getrübt wird (da das Produkt in Ethanol 90 % nicht völlig unlöslich ist, wird die Chloridprobe nie ganz negativ) und getrocknet.
Ausbeute: 2,45 g (82 %) hell orangefarbenes Kristallpulver; 1 Teil löst sich in 96 Teilen Wasser von 19 °C

Abb.: trans-Tetraammindinitrokobalt(III)-chlorid
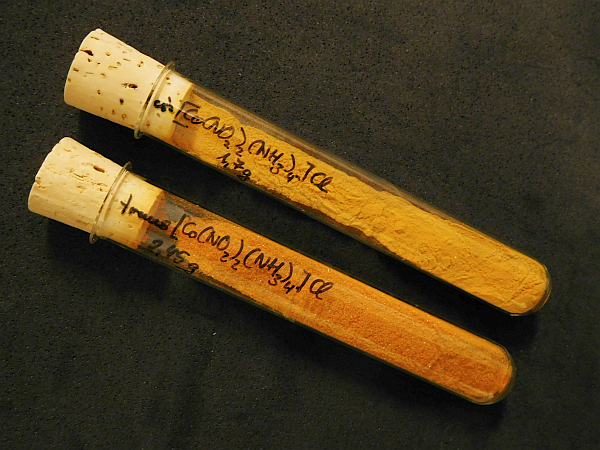
Abb.: cis- und trans-Tetraammindinitrokobalt(III)-chlorid
12. mer-Triammintrinitrokobalt(III) (Kobalttriamminnitrit nach Erdmann und Gibbs)
(nach S.M. Jörgensen; Z. f. anorgan. Chemie 17(1898): 475)
[Co(NH3)3(NO2)3] – Molmasse: 247,9 g/mol
In 80 ml Wasser löst man kalt 10 g Ammoniumchlorid und 13,5 g Natriumnitrit, fügt 40 ml Ammoniaklösung 25% und schließlich eine Lösung von 9 g Kobalt(II)-chlorid-Hexahydrat in 20 ml Wasser zu. Die dunkelrote Flüssigkeit bekommt rasch einen Braunstich und färbt sich beim anschließenden Durchleiten von Luft während 4 Stunden tief dunkelbraun. Man gießt sie in eine große Abdampfschale und stellt sie in den Zug (s. Vorbemerkung!). Die Weiterverarbeitung soll nach 72 Stunden erfolgen. Bei meinem Versuch hatten sich schon nach 48 Stunden eine große Menge große, dunkel-rotbraune Kristalle neben einem feineren, braungelben Kristallpulver ausgeschieden, und die Flüssigkeit war hell-braungelb geworden. Das Rohprodukt wird abgesaugt und mit 5 x 40 ml kaltem Wasser gewaschen. Erhalten wurden 7,5 g rotbraune Kristalle vermischt mit einer kleinen Menge einer feinkristallinen, gelben Substanz (vermutlich trans-Tetraammindinitrokobalt(III)-chlorid).


Abb: Rohprodukt, oben in der Reaktionsmischung ausgefallen, unten nach Absaugen und Waschen
Das Rohprodukt wird umkristallisiert, indem man es mit 50 ml 1 %iger, heißer (ca. 80° C) Essigsäure übergießt, gut verrührt und absaugt. Dies wird wiederholt, bis sich die braunrote Substanz gelöst hat und nur noch ein wenig gelbbrauner Rückstand übrig geblieben ist, wozu ca. 150 ml nötig sind. Die vereinigten Filtrate lässt man abkühlen, saugt das gelbbraune, feinkristalline Produkt ab und wäscht mit wenig kaltem Wasser nach.
Ausbeute 5,8 g (62 %), kurkumafarbenes, feines Kristallpulver

Abb.: Triammintrinitrokobalt(III)
13. Ammonium- und Kalium-trans-diammintetranitrokobaltat(III) (Erdmann'sches Salz)
(nach S.M. Jörgensen; Z. f. anorgan. Chemie 17(1898): 476-479)
NH4[Co(NH3)2(NO2)4] – Molmasse: 294,9 g/mol
K[Co(NH3)2(NO2)4] – Molmasse: 316 g/mol
In 80 ml Wasser löst man kalt 10 g Ammoniumchlorid und 13,5 g Natriumnitrit und fügt 2 ml Ammoniaklösung 25% sowie eine Lösung von 9 g Kobalt(II)-chlorid-Hexahydrat in 20 ml Wasser zu. Man leitet 1½ Stunden Luft durch die Mischung und stellt die resultierende, gelbbraune Flüssigkeit in einer flachen Abdampfschale für mehrere Tage in den Zug. Ich habe das Experiment ohne Ventilator durchgeführt. Nach 6 Tagen hatten sich reichlich rotbraune Kristalle, vermischt mit einem gelbbraunen Pulver, abgesetzt. Man rührt gut durch, und gießt die trübe, gelbbraune Flüssigkeit durch ein Filter von den schwereren rotbraunen Kristallen ab. Die Kristalle (4 g) werden beiseite gestellt und das Filtrat (100 ml) erneut dem Zug ausgesetzt. Nach 48 Stunden - diesmal vor dem Ventilator - hatten sich eine weitere Portion Kristalle abgeschieden, die wie oben abgeschlämmt werden. Die restliche Mutterlauge wird verworfen.
Das Rohprodukt, im ganzen 6,2 g, wird in einem Becherglas mit 50 ml Wasser von 45 °C übergossen und geschwenkt, bis nichts mehr in Lösung geht. Die Lösung wird filtriert und der Rückstand erneut mit warmem Wasser ausgezogen bis nur noch ein geringer, gelbbrauner Bodensatz übrig bleibt (bei mir waren zwei Lösungsvorgänge ausreichend). Die vereinigten Filtrate stellt man in einer Abdampfschale auf die Heizung und lässt sie langsam eindunsten.
Ausbeute: 5,3 g (47,3 %) dunkel-braunrote Kristalle; leicht wasserlöslich
Anmerkung: nach mehrfacher Durchführung dieser Synthese kann ich folgendes ergänzen: am besten arbeitet man bei niedrigen Temperaturen (im Schuppen bei 10 °C hatte ich die höchste Ausbeute) und lässt vor dem Ventilator solange eindunsten (binnen 48-72 Stunden) bis im Bodensatz einzelne weiße Kochslzkristalle erkennbar werden. Dann gibt man etwas Wasser zu, bis sich das Kochsalz wieder löst und schlämmt von den rotbraunen Kristallen wie oben ab. Am besten wäscht man diese vor dem Lösen in warmem Wasser mit ein wenig gesättigter Lösung des Produktes (aus einer früheren Darstellung). Beim langsamen Eindunsten kristallisiert das Präparat in schönen, kurzen, säulenförmigen Kristallen aus.
Das Kaliumsalz wird erhalten, indem man 4 g des Ammoniumsalzes unter Erwärmen in 25 ml Wasser löst und mit einer Lösung von 2 g Kaliumacetat in 10 ml Wasser vermischt. Beim Abkühlen scheiden sich ziemlich große Kristalle aus, die abgesaugt und auf der Fritte einmal mit 5-7 ml eiskaltem Wasser und 3 x 10-15 ml Ethanol gewaschen werden. Die Mutterlauge lässt man auf der Heizung auf etwa die Hälfte eindunsten, wobei sich nochmals etwas Kaliumdiammintetranitrokobaltat(III) abscheidet, das ebenfalls gewaschen und getrocknet wird.
Ausbeute: 3,6 g (83 %) dunkelbraune Kristalle, mäßig schwer wasserlöslich

Abb.: Ammonium- und Kalium-diammintetranitrokobaltat(III)
14. Natrium-hexanitrokobaltat(III)
(nach E. Biilmann; Z. f. analytische Chemie 39(1900): 286-287)
Na3[Co(NO2)6] – Molmasse: 403,9 g/mol
Man erhitzt 15 ml Wasser bis fast zum Sieden und gibt 15 g Natriumnitrit (welches sich nicht ganz auflöst) sowie 5 g Kobalt(II)-nitrat-Tetrahydrat zu. Nach dem Erkalten fügt man portionsweise (jede Minute 1 ml) insgesamt 5 ml 50 %ige Essigsäure zu, wobei kleiner Mengen nitroser Gase entweichen, und leitet dann für ½ Stunde einen kräftigen Luftstrom durch die tiefbraune Mischung. Nach zweistündigem Stehenlassen findet man einen gelbbraunen Bodensatz und darüber eine dunkelbraune Flüssigkeit vor. Man gießt durch ein Filter ab, löst den Bodensatz unter Erwärmen in 5-10 ml Wasser und gibt die Lösung ebenfalls auf das Filter. Das vereinigte Filtrat (etwa 35 ml) wird nach dem Abkühlen portionsweise mit insgesamt 35 ml absolutem Ethanol versetzt. Es fällt ein orangefarbener Niederschlag aus, der nach einstündigem Stehen abgesaugt und mit 3 x 5-10 ml Ethanol sowie 2 x 5-10 ml Ether gewaschen und getrocknet wird.
Ausbeute: 5,6 g (80,4 %) leuchtend orangefarbenes Pulver; sehr leicht wasserlöslich

Abb: Natrium-hexanitrokobaltat(III)
Entsorgung:
Die verschiedenen anfallenden Mutterlaugen und Filterrückstände werden als anorganischer Sondermüll entsorgt. Auch nicht mehr benötigte Präparate kommen in den Schwermetallabfall.
Erklärungen:
(Im Seminar bei Professor Jørgensen, Winter 1892/93, chemisches Institut der Universität Kopenhagen)
Reiseleiter: Herr Jørgensen …
Jørgensen: (hebt eine Augenbraue) Professor Jørgensen, wenn ich bitten darf!
Reiseleiter: Herr Professor Jørgensen! Ihre Synthesen unterscheiden sich oft nur in Details. Wie sind sie auf diese Vorschriften gekommen?
Jørgensen: Im Verlaufe der vielen Jahre, in denen ich mit Kobaltammoniaksalzen arbeite, habe ich selbstverständlich verschiedene Erfahrungen über die bequemsten und ergiebigsten Darstellungsweisen gesammelt. Ich habe dazu eine große Reihe von Versuchen angestellt. Als Bespiel mag ihnen die Darstellung der verschiedenen Nitrokobaltammoniakate dienen. Ich habe den Prozeß unter jedesmal wenig abgeänderten Bedingungen genauer untersucht. Unter Einsatz immer der gleichen Menge Ammoniumchlorid und Natriumnitrit sowie Kobaltchlorür spielen sich, je nach der Ammoniamkenge, folgende Reaktionen ab:
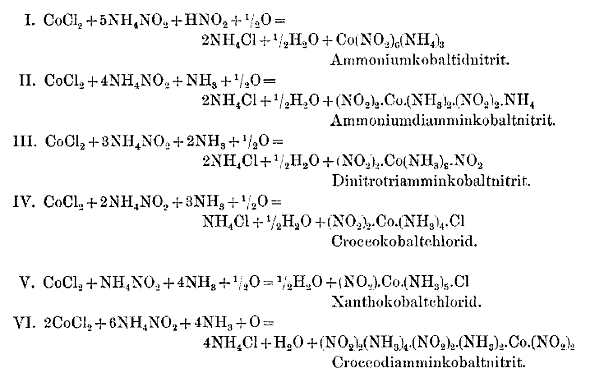
Die Reaktion VI ist nur eine Kombination von II und IV. Unschwer lässt sich bei zweckmäßiger Reaktion der Ammoniakmenge das eine oder andere Reaktionsprodukt in weit überwiegender Menge erhalten. Weniger als 5 mol Ammoniumnitrit (aus Ammoniumchlorid und Natriumnitrit im Ansatz dargestellt – Anmerkung der Reiseleitung) auf 1 Atom Co (was den oben angegebenen Gewichtsmengen annähernd entspricht) lassen sich mit Vorteil nicht verwenden, weil sich sonst größeren Mengen des Kobalts als grünes Oxyduloxydhydrat ausscheiden.
Die Darstellung reinen Hexaamminkobaltchlorids hat mir dagegen Schwierigkeiten bereitet. Ich habe hauptsachlich das 1868 von E. J. Mills angegebene Verfahren angewendet, nämlich Erhitzen des Chloropentamminchlorids mit Ammoniakflüssigkeit in einer verschlossenen Sodawasserflasche. Ich verwende hierzu mit sehr gutem Erfolg Sodawasserflaschen mit Patentverschluß von Fa, Siemens in Dresden. Sie vertragen sehr gut Erhitzen mit 20%iger Ammoniakflüssigkeit im Wasserbade. Die Flasche wird ganz in Leinwand gehüllt und wird in dem kalten Wasserbad in einen großen Zinkcylinder gestellt. Herr Cand. Dorph Broager hat eine ganze Reihe Versuche nach diesem Verfahren ausgeführt. Es zeigte sich, daß die Menge des zugesetzten Salmiaks kritisch ist. Die besten Resultate hat mein Assistent mit 8 g Salmiak auf … (wühlt in seinen Papieren) … 10 g Purpureochlorid …
Reiseleiter: (schnell) Ähm, sehr interessant Herr Professor, sie haben ja wirklich systematisch verschiedene Synthesen durchprobiert. Ihre Anleitungen sind exzellent! Nur beim Präparat Nummer 8 sind unsere Ausbeuten hinter den Erwartungen zurückgeblieben. Nach ihrem Artikel wären bei der Darstellung des Sulfates knapp 70% Ausbeute zu erwarten. Unser Versuch hat nur etwa die Hälfte ergeben. Aber sie wollten uns die Bildung der verschiedenen Salze erklären…
Jørgensen: Haben sie sich denn genau an die Vorschrift gehalten?
Reiseleiter: Hmm! Eigentlich schon. Beim Eindampfen des Chlorids im ersten Syntheseschritt sind wir etwas über das Ziel hinausgeschossen und haben auf 80 ml eingedampft. Nach dem Zusatz von Salzsäure fiel schon vor dem Erwärmen ein Niederschlag aus. Das Rohprodukt hat sich nur zu etwa ¾ in der angegebenen Menge Wasser gelöst
Jørgensen:: Das ist normal. Das Präsochlorid, fast das gesamte Purpureochlorid und ein Teil des Tetraamminchlorosalzes bleiben ungelöst.
Reiseleiter: Und dann haben wir keine Ammoniumsulfatlösung 1:5 vorrätig gefunden, von der wir ¼ des Volumens hätten zugeben müssen. Stattdessen haben wir eine gesättigte Lösung von der abgeschätzten Menge Ammoniumsulfat zugesetzt. Aber das sollte doch nicht so ins Gewicht fallen?
Jørgensen: Möglicherweise haben sie durch das zu starke Eindampfen einen erhöhten Anteil an Pentaamminpurpureochlorid in ihrer Rohsubstanz gehabt. Haben sie die Mutterlauge daraufhin überprüft?
Reiseleiter: Äh, nein! Auf die Idee bin ich nicht gekommen. Und von der Rohsubstanz ist nichts mehr übrig.
Jørgensen: (herablassend) Sie müssen lernen analytisch zu denken! Von jedem Präparat muss etwas zurückgelegt werden. Und was das präparative Arbeiten angeht brauchen sie wohl einfach mehr Erfahrung!
ein Besucher: (merkt, daß das Gespräch ungemütlich zu werden droht) Herr Professor, sie wollten uns noch die Bildung der anderen Salze erklären.
Jørgensen: (guckt scharf) Einen Moment noch! Die von ihnen angewandte Methode mit der Aktivkohle zur Darstellung des Luteokobaltchlorids war mir nicht bekannt. Vermutlich agiert die Kohle als Katalysator. Haben Sie das entdeckt?
Besucher: (verlegen) Die Methode ist nicht von uns. Herr Bjerrum hat sie 1941 veröffentlicht.
Jørgensen: (etwas irritiert) Sie meinen wohl 1841? Bjerrum? Ich habe einen Studenten dieses Namens, aber der kann das ja nicht gewesen sein... Hätten sie die Freundlichkeit mir das Zitat mitzuteilen?
Besucher: (schwitzt) Sicher doch, ich suche es ihnen in den nächsten Tagen heraus ... Übrigens enthält das nach dieser Vorschrift hergestellte Hexaamminkobalt(III)-chlorid noch etwas Pentaamminchlorokobalt(III)-chlorid und …
Jørgensen: (unterbricht den Besucher) Junger Mann! Was sind denn das für unnötig komplizierte Bezeichnungen? Sagen sie Luteokobaltchlorid und Purpureokobaltchlorid und jeder wird Sie verstehen! Und daß Kobalt dreiwertig ist versteht sich auch von selbst.
Reiseleiter: (seufzt halblaut) Ach ja, ständig diese neuen Nomenklaturregeln …!
Murmeln und verhaltene Heiterkeit in der Reisegruppe. Einige Mitreisende machen grinsend eine Kopfbewegung hin zum Reiseleiter.
Jørgensen: Aber die Verunreinigung können Sie aus ihrem Produkt einfach entfernen. Sie müssen es nur aus möglichst wenig 5 %igem Ammoniakwasser umkristallisieren. Obwohl sich dabei immer ein wenig Salz zersetzt, können sie über 90 % reines Hexamminchlorid zurückgewinnen.
Besucher: (erleichtert) Vielen Dank für den Rat! Übrigens: Wie prüfen Sie die Reinheit ihrer Präparate?
Jørgensen: Zum einen durch Analyse, indem wir die gefundenen Werte mit den aus der Formel berechneten vergleichen. Kobalt bestimmen wir als Kobaltsulfat, indem die Substanz verglüht und der Rückstand mit Schwefelsäure abgeraucht wird. Ammoniak wird wie üblich durch starke Laugen ausgetrieben und dann titriert. Chlor wird im Rückstand als Chlorid mit Silbernitrat ausgefällt und gravimetrisch bestimmt. Nitrit wird mit übermangansaurem Kali titriert.

Daneben geben viele der Kobaltammoniakate specifische Niederschläge mit bestimmten Reagentien. Zum Beispiel können sie ihr Luteokobaltchlorid auf Spuren von Roseosalz überprüfen, indem sie es in verdünntem Ammoniak lösen und dann mit Ammoniumoxalat fällen. Noch 0,3% Beimengung von Chloro- oder Aquopentamminsalz machen sich im Filtrat durch eine Rotfärbung kenntlich. Das Croceokobaltchlorid darf mit Ammoniumoxalat auch nach längerem Stehen keinen Neiderschlag geben, der von nicht ausreichend ausgewaschenem Xanthokobaltsalz herrühren würde. Chloropurpureochlorid und die Roseosalze können sie mit Natriumpyrophosphat auf Verunreinigung mit Luteosalz prüfen.

Abb.: Reaktion mit Quecksilberchlorid (v.l.n.r.: Hexaamminkobalt(III)-chlorid, Pentaamminnitrokobalt(III)-chlorid, Pentaamminaquokobalt(III)-chlorid), Pentaamminchlorokobalt(III)-chlorid, cis- und trans-Tetraammindinitrokobalt(III)-chlorid, Trinitrotriamminkobalt-(III)]
ein anderer Besucher: Und wie steht es um die Bildung der anderen Komplexe?
Jørgensen: Ich muss sie doch sehr bitten, sich einer genauen Ausdrucksweise zu befleißigen! Wir sprechen hier von Kobaltammoniakaten, komplexe Verbindungen sind solche, die mehr als ein Atom Kobalt im Molekül enthalten.
Die Carbonatotetramminkobaltsalze hat Vortmann schon 1877 und 1882 beschrieben. Ich habe lediglich ihre Konstitution aufgeklärt. Die Darstellung geschieht verhältnismäßig leicht, indem man ammoniakalische Kobaltlösung mit einem großen Überschuss an Ammoniumcarbonat oxydiert und dann eindampft. Auf letztere Weise werden besonders das Nitrat, das Sulfat, das Bromid und das Jodid leicht und in reichlicher Menge beim Auskrystallisieren aus den langsam erkalteten Lösungen erhalten, das Chlorid dagegen muß durch Weingeist aus der passend eingeengten Lösung ausgeschieden werden.
Die Darstellung von reinen Pentaaminchlorokobaltchlorid wurde in meinem Institut von Sørensen ausgearbeitet. Frühere Darstellungsweisen, waren langwierig. Rose hatte 1871 die Verhältnisse ausführlich studiert. Um eine einigermaßen reichliche Ausbeute zu erreichen, mußte er 20 Stunden Luft durch die Lösung leiten und sie dann drei Monate in einem nicht verschlossenen Kolben stehen lassen. Hier betrug die Ausbeute an rohem, gemischtem Salz etwa 90 % von der theoretischen. Ich schlug Sørensen vor, Purpureochlorid mit Tetramminsalz als Durchgangsglied darzustellen. Der Weg, den wir hier beschritten, beinhaltet zunächst eine Oxydierung, bei der vorzüglich Pentaaminroseosalz [Pentaamminaquokobalt(III)-chlorid – Anm. der Reiseleitung] neben Tetraaminkarbonatokobaltchlorid gebildet wird. Letzteres wird beim Eindampfen mit Salmiak in Pentaamminchlorid überführt. Die Umwandlung ist aber, wie die Untersuchungen zeigten, bis hierhin noch nicht vollständig. Solange noch Spuren von Ammoniumkarbonat in der Lösung sind, bleibt etwas Carbonatotetraamminsalz zurück. Man darf aber nicht ganz eindampfen, da sich das Präparat sonst unter Bildung von Kobaltchlorür [Kobalt(II)-chlorid - Anm. der Reiseleitung] zersetzt. Sørensen hat diese Zersetzung durch Zusatz von mehr Ammoniak zu verhindern gesucht. Dabei wird aber in erheblicher Menge Luteosalz gebildet, was auch nicht wünschenswert ist. Die oben gegebene Anleitung führt zu guten Ausbeuten. Übrigens können sie aus der Farbe der Mutterlauge am Ende schließen, wie der Proceß verlaufen ist. Die Flüssigkeit kann fast farblos sein oder grün. Diese beiden Farben deuten eine gute Ausbeute an, wogegen eine stärkere blaue Farbe auf einen stärkeren Gehalt yon Kobaltchlorür deutet, besonders, wenn die Farbe sich auch nach dem Erkalten blau erhält.
Die Darstellung des Purpureokobaltchlorids ist, wie sie wissen, von großer Bedeutung, weil sie es uns erlaubt, das Kobalt völlig vom Nickel abzutrennen, welches kein solches Salz bildet. Das Atomgewicht des Kobalts ist jetzt zweifelsfrei zu 58,9 bestimmt worden und damit tatsächlich etwas größer als die des Nickels. Übrigens ist Purpureochlorid in guter Qualität rein im Handel erhältlich und dient als Ausgangssubstanz für weitere Darstellungen. So erleidet es durch Hydrolyse in schwacher Ammoniaklösung einen Austausch des an das Kobalt gebundenen Chlors durch Wasser und man erhält das Roseokobaltchlorid, das man aber in der Kälte abscheiden muß. Durch Erhitzen mit Salzsäure wird das Purpureosalz zurückgebildet. Das Oxalat ist in der Darstellung viel weniger kritisch und auch stabiler als das Roseochlorid.
Besucher: Wie erklärt es sich, daß sich aus dem Pentaamminchlorokobalt(III)-chlorid (der Professor hebt eine Augenbraue) … äh, ich meine, aus dem Purpureokobaltchlorid, nur zwei Drittel des Chlors als Silberchlorid fällen oder durch Schwefelsäure abspalten lassen?
Jørgensen: In diesen Salzen kommt die Gruppe –NH3 –NH3 –NH3 –NH3 vor, welche in Verbindung mit einem Äquivalent des Kobalts ein Ammonium bildet, von ähnlichem ungemein stark elektropositivem Charakter wie das Tetraäthylammonium. Es scheint dann ganz natürlich, daß die zwei anderen Aequivalente Kobalt, die im trivalenten Kobaltatom unzertrennlich mit dem erst en verbunden sind, von jener stark elektropositiven Atomgruppe. stark influirt werden müssen und selbstverständlich in elektronegativer Richtung. Bezüglich dieser zwei Valenzen zeigt sich das Kobaltatom daher in diesen Verbindungen als ein weit elektronegativeres Element, als wir es sonst kennen. Diesen Verbindungen kommt somit folgende Konstitution zu (wo a ein Ammoniakmolekül darstellt):

Das direkt an das Metallatom gebundene Chlor ist fest gebunden, nicht ionisierbar und nicht durch Silbernitrat auszufällen. Luteokobaltchlorid kann also drei, Puprpureokobaltchlorid zwei und das Präsochlorid nur eines der enthaltenen Chloratome abspalten, nämlich dasjenige, das an die Ammoniak-Kette gebunden ist.
Besucher: Und die Nitroamminkobaltsalze?
Jørgensen: Diese Reihe ist sehr interessant, denn sie zeigt, daß wir in den Ammoniakaten den Ammoniak succesive durch Säurereste substituiren können.

Abb.(v.l.n.r.): Hexaamminkobalt(III)-chlorid, Pentaamminnitrokobalt(III)-chlorid, cis- und trans-Tetraammindinitrokobalt(III)-chlorid, Triammintrinitrokobalt(III), Kaliumdiammintetranitrokobaltat(III) und Natriumhexanitrokobaltat(III)
Besucher: (provokativ) Aber es gibt doch zwei verschiedene Dinitrotetraamminkobaltsalze.
Jørgensen: In der Tat kennen wir zwei solche Verbindungen! Die Croceosalze wurde 1875 von Gibbs beschrieben. Die damit isomeren Flavosalze habe ich entdeckt, als ich Carbonatotetraamimminkobaltnitrat mit Natriumnitrit umsetzte, in der Absicht Croceosalz darzustellen. Die Verbindungen unterscheiden sich schon äußerlich in der Farbe, aber auch in ihrer Löslichkeit und manchen Fällungsreaktionen. Flavokobaltchlorid wird durch Quecksilberchlorid als Doppelsalz gefällt, Croceokobaltchlorid nicht.
Besucher: Wie müssen wir uns die Isomerie vorstellen?
Jørgensen: Das ist nur so zu erklären, daß die vom Kobalt betätigten Valenzen verschieden sein müssen. Ich bezeichne sie mit alpha, beta und gamma.
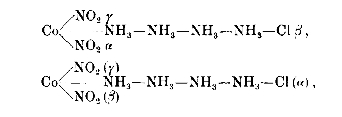
Genau so, wie ich es für die Ethylendiaminverbidungen des Kobalts gefunden habe. Es existieren zwei Verbindungen der Formel Coen2Cl2. Die eine dieser Verbindungen ist, wie das Präsokobaltchlorid, dunkelgrün, die andere tiefviolett gefärbt. Ich nenne sie das Violeosalz. Ihre Konstituion ist:
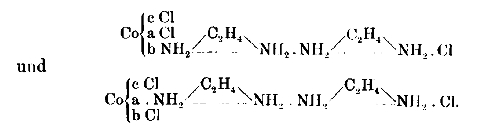
Sie bemerken die vollständige Analogie zu den Ammoniakverbindungen. Die zweigliedrige Äthylendiaminkette ersetzt die viergliedrige Ammoniakkette!
Besucher: Und die viergliedrige Ammoniakkette bleibt immer erhalten?
Jørgensen: Sie ist jedenfalls sehr stabil und bleibt bei vielen Umsetzungen erhalten. So können, ausgehend von den Carbonatotetraamminkobaltsalzen, leicht und in guter Ausbeute andere Tetraamminverbindungen erhalten werden, indem sich die Carbonatgruppe durch Säuren abspalten lässt und dann die Säurereste oder auch Wassermoleküle an deren Stelle sich an das Kobalt binden. Sie haben auf diese Weise Diaquoroseosalze und Präsokobaltchlorid dargestellt. Ein schlagender Beweis für das Vorhandensein der viergliedrigen Ammoniakkette in den Tetraamminsalzen ist folgender: Verreiben sie irgendein Tetraamminsalz - Chlorotetraamminchlorid, Diaquoroseosalz, Flavosalz oder Croceosalz - mit Schwefelsäure, geben sie etwas konzentrierte Salzsäure zu und lassen sie das ganze einen Tag stehen. In jedem Fall werden sie finden, daß sich saures Präsokobaltsulfat, also ein Dichloro-Tetraamminkobaltsalz, bildet.
Aber wir kennen natürlich auch Tri- und Diamminverbindungen. Das Triamminnitrit von Gibbs und Erdmann beispielsweise. Darin ist die Ammoniakkette nur dreigliedrig:
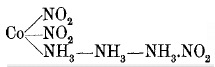
Und das Kaliumdiamminkobaltnitrit müssen wir uns so gebaut vorstellen:
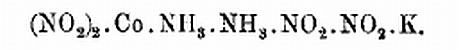
(verschiedene Besucher runzeln die Stirn, sagen aber aus Höflichkeit nichts)
Reiseleiter: Sie haben uns gezeigt, daß beim Behandeln mit Schwefelsäure und Salzsäure sowohl aus den Croceokobaltsalzen als auch aus den Flavokbaltsalzen Präsosalze entstehen. Sie haben ausserdem aber gefunden, dass, wenn man kleine Mengen Flavokbaltchlorid und Croceokobaltchlorid mit etwas konzentrierter Salzsäure kurz erwärmt und dann stehen lässt, die Reaktionen verschieden sind. Im ersten Fall erhäklt man einen grünen Niederschlag von Präsokobaltchlorid, im zweiten aber einen roten Niederschlag, der aus dem von Ihnen entdeckten Chloronitrotetraamminkobaltchlorid besteht. Wie erklärt sich das verschiedene Verhalten der Salze?

Abb.: Reaktion von cis-Tetraammindinitrokobalt(II)-nitrat (Flavosalz - links) und dem trans-Isomeren (Croceosalz - rechts) nach kurzem Erhitzen mit konzentrierter Salzsäure
Jørgensen: Dieses Verhalten stützt meine Theorie: die Valenzen sind verschieden! Im Croceosalz bedarf es zur Abspaltung einer der beiden Nitrogruppen besonders energischer Reagentien. Das Flavosalz gibt beide Nitrogruppen gleich leicht ab.
ein Besucher: Wenn es aber zwei isomere Reihen von Dinitrotetraamminkobaltsalzen gibt, müsste es dann nicht auch zwei Dichlorotetraamminkobaltsalze geben, also ein Isomeres zum Präsokobaltchlorid?
Jørgensen: Das habe ich schon in meinen früheren Veröffentlichungen nicht ausgeschlossen. Bei den Dichloro-diäthylendiammminkobaltsalzen kennen wir ja Präso- und Violeosalze. Aber bei den Tetraamminkobaltverbindungen ist ein Violeosalz nie beobachtet worden.
Besucher: Das ist reichlich kompliziert. Müsste es dann nicht auch zwei Triamminkobaltnitrite geben?
Jørgensen: Das ist Spekulation! Wir müssen von den experimentellen Tatsachen ausgehen und die sind eindeutig. Die Salze von Gibbs und Erdmann sind in ihrer Kristallform und ihrem chemischen Verhalten identisch. Es ist nur ein Dichlorotetraamminkobaltchlorid und nur ein Triamminkobaltnitrit bekannt.
Reiseleiter: Sie nennen das Salz von Erdmann und Gibbs “Triamminkobaltnitrit“. Sie gehen also davon aus, dass diese Verbindung beim Lösen in Wasser in ein positiv geladenes Triammindinitrokobalt-ion und ein Nitrition zerfällt?
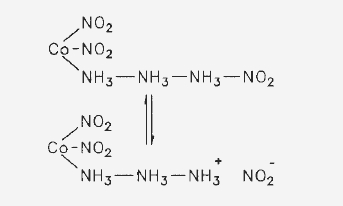
Jørgensen: es kann nicht anders sein, denn das Kobaltatom hat keine weiteren Valenzen, an die es den dritten Säurerest binden könnte. Weil es aber etwas eigentümlich erscheint, daß das Nitrit weit schwerer löslich als die anderen Salze ist, so ist es allerdings möglich, daß es als
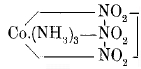
aufzufassen ist.
Die Unruhe in der Reisegruppe, angesichts der immer phantastischer werdenden Strukturformeln, nimmt zu. Wir wollen uns lieber von dem distinguierten Professor Jørgensen verabschieden und in die Gegenwart zurückkehren. Seine letzte Bemerkung aber sollten wir gut im Gedächtnis behalten! Aus einer Hypothese Voraussagen abzuleiten, die sich später experimentell bestätigen, ist jedes Mal ein grandioser Erfolg. Mendelejew hatte aus seinem Periodensystem die Existenz noch unbekannter Elemente sowie deren Eigenschaften vorausgesagt. Gallium und Germanium bestätigten seine Hypothese glänzend. Jørgensen ist sich sicher “daß die Ammoniakgruppen in den Kobaltbasen sich gegenseitig binden … besonders nachdem ich es für die Platinbasen bewiesen habe“. Sein System ist gut begründet und ohne Widersprüche. Oder doch nicht? Dazu wird sich in Kürze ein anderer Wissenschaftler zu Wort melden, den wir im zweiten Teil der Reise besuchen werden. Die Reiseleitung hofft, daß ihr eine interessante Zeit hattet!
Hommage und Nachbemerkung:
Wie wir heute wissen, lag Sophus Mads Jørgensen mit seiner Strukturtheorie "Zur Constitution der Cobaltbasen" falsch. Dennoch sollten wir seine Arbeit honorieren. Heute weitgehend vergessen, war er zu seiner Zeit ein international hochgeschätzter und anerkannter Wissenschaftler. Er hat mit in jahrzehntelangen systematischen Experimenten die meisten jener Syntheseanleitungen ausgearbeitet, denen wir hier gefolgt und die bis heute unübertroffen sind. Er hat die Zusammensetzung der Kobaltverbindungen durch sorgfältige Analyse genau aufgeklärt. Ironischerweise haben einige seiner Präparate später wesentlich zum Sturz der von ihm vertretenen Strukturlehre beigetragen, doch davon das nächste Mal mehr! Für seine Verdienste zeichnete die französische Academie des Sciences Jørgensen 1906 mit der Lavoisier-Medaille aus. Fast alle Vorschriften in der Sekundärliteratur, die dem Autor dieses Artikels bekannt sind, haben direkt - teils wörtlich! - seine Anleitungen übernommen. So auch in der letzten Ausgabe des "Jander/Blasius; Anorganischen Chemie“ von 2012. Lediglich die Darstellung des Hexaamminkobalt(III)-chlorids stammt aus einer anderen Quelle, nämlich vom Sohn eines von Jørgensens Schülern!
In diesem Zusammenhang erlaubt sich die Reiseleitung noch eine Bemerkung: In die Vorschriften des Jander/Blasius haben sich Fehler eingeschlichen!
- Zum Auskristallisieren des cis-Tetraammindinitrokobalt(III)-chlorids wird die Zugabe von 10 ml Ethanol zur Reaktionsmischung angegeben, wo Jørgensen 100 ml angewandt hatte – und wie die oben berichteten Versuche zeigen, ist diese Menge auch notwendig, um eine Fällung des Salzes zu erzielen. Hatte der Redakteur womöglich eine untergründige Abneigung gegen die Ver(sch)wendung solcher Mengen Alkohol?
- Und bei der Synthese des trans-Tetraamminndinitrokobalt(III)-chlorids wird gefordert “mit einer wässrigen Lösung von 4 g Ammoniumchlorid“ zu fällen, während Jørgensen eindeutig schreibt "man versetzt das klare und heiße Filtrat sogleich mit 4 g in Wasser klar löslichem Salmiak“. Man muss davon ausgehen, daß die Ausbeute durch diese Änderung stark leidet.
Solche Fehler zeigen, daß hier nur abgeschrieben, aber nicht selbst experimentiert wurde. Zu den vielen Eindrücken der Reise sollten wir auch mitnehmen, daß es sich lohnt, Versuchsbeschreibungen im Original zu lesen. Geschrieben von jemandem, der die Versuche auch wirklich gemacht hat!
Literatur:
Fast alle von S.M. Jørgensen verfassten Artikel sind bei identischem Titel (“Beiträge zur Chemie der Kobaltammoniakverbindungen“ und vor allem “Zur Konstitution der Kobalt-, Chrom- und Rhodiumbasen“) fortlaufend nummeriert und oft so umfangreich, daß ich in den oben gegebenen Referenzen die Titel weggelassen und nur die Seiten angegeben habe, auf denen die Synthese der betreffenden Substanz beschrieben ist.
Viele Synthesen finden sich in dem Artikel “Zur Darstellung der Kobaltammoniaksalze“; Zeitschrift für anorganische Chemie 17 (1898) zusammengefasst. Dort wird weiter auf ausführliche Syntheseanleitungen an anderer Stelle verwiesen, die überwiegend in der selben Zeitschrift, manche auch in der Vorgängerzeitschrift, dem Journal für praktische Chemie [2] (es gibt eine Reihe [1] und eine Reihe [2] mit identischer Nummerierung der Ausgaben!) veröffentlicht wurden.
Die von Jørgensen erwähnte Darstellungsweise des Hexaamminkobalt(III)-chlorids geht zurück auf:
Mills EJ: On certain Cobaltamines; The London, Edinburgh and Dublin Philosophical Magazine and Journal of Science Fourth Series, No. 35, April 1868: 245-261. Die Darstellungsweise nach Jannik Bjerrum findet sich in dessen Habilitationsschrift, die er 1941 unter der deutschen Besatzung anfertigte: Metal Ammine Formation in Aqueous Solution; Copenhagen 1941, S.241. Jannik Bjerrum (1909-1992) war der Sohn von Jørgensens Schüler Niels Janniksen Bjerrum (1879-1958) und ebenfalls Chemieprofessor in Kopenhagen.
Die beste Darstellungsmethode für Pentaamminchlorkokobalt (III)-chlorid wurde von Jørgensens Assistent Søren Peter Lauritz Sørensen (1868-1939) ausgearbeitet und in der Zeitschr. f. anorgan. Chemie 5(1894):354-373 unter dem Titel "Kritische Präparatenstudien“ veröffentlicht. Sørensen war vornehmlich Analytiker. Er ist vor allem durch die Definition des pH-Wertes bekannt geworden. Aber auch seine Darstellungsvorschriften haben ihn überdauert. Besonders reine Analysenchemikalien wie Natriumdihydrogenphosphat und Natriumoxalat "nach Sörensen“ geben davon ein Zeugnis ab.
Die ersten Veröffentlichungen über Triammintrinitrokobalt(III) finden sich bei:
O.L. Erdmann: Ueber einige salpetrigsaure Nickel- und Kobaltverbindungen; J. f. prakt. Chemie[1] 97, 1866: 412-413 und
Wolcott Gibbs; Researches on the Hexatomic Compuounds of Cobalt; Proceedings of the american academy of arts and sciences Vol X (1874), I: 1-38
Der Artikel von Einar Christian S. Biilmann (1873-1946) (Ueber die Darstellung des Natriumkobaltidnitritis und seine Anwendung zum Nachweis von Kalium; Z. f. analytische Chemie 39(1900): 284-289) ist zwar erst lange nach unserem Besuch im Jahre 1892 erschienen, aber da auch er ein Schüler Jørgensens war, bitte ich, mir diesen Vorgriff in der Zeit zu verzeihen. Auch Biilmann hatte von 1907-1943 einen Lehrstuhl an der Universität Kopenhagen inne, allerdings den für Organische Chemie.
In der Sekundärliteratur finden sich die Synthesevorschriften Jørgensens unter anderem in:
Biltz H und Biltz W: Übungsbeispiele aus der unorganischen Experimentalchemie, 2. Auflage; Leipzig, Verlag von Wilhelm Engelmann 1913: Seiten 162-173
Brauer G: Handbook of Preparative Inorganic Chemistry, Vol II, Second Edition; Academic Press London-New York 1963: Seiten 1531-1541
Hecht H: Präparative Anorganische Chemie, Springer-Verlag OHG, Berlin/Göttingen/Heidelberg 1951: Seiten 163-182
Schweda E: Jander/Blasius Anorganische Chemie II, quantitative Analyse und Präparate, 16. Auflage; Hirtzel-Verlag Stuttgart 2012 (ISBN 978-3-7776-2133-3): Seiten 338-341
Vanino L: Handbuch der präparativen Chemie; Verlag von Ferdinand Enke, Stuttgart 1913; Band 1: Seiten 535-541
Die Äußerungen Jørgensens sind teilweise wörtlich aus seinen Artikeln - und zwar auch aus späteren, bis 1899 erschienen Artikeln - entnommen, teilweise sinngemäß frei formuliert. Eine Kennzeichnung jedes einzelnen Zitates hätte den Text unleserlich gemacht. Hinweise auf persönliche Eigenarten und die wissenschaftliche Ausrichtung Jørgensens habe ich den Artikeln von George B. Kauffmann:
"Sophus Mads Jørgensen and the Werner-Jørgensen-Controversy"; Chymia 6 (1960): 180-204 und
"Sophus Mads Jørgensen, a chapter in coordination chemistry history", Journal of chemical Education 36 (1959): 521-527
entnommen. Aus letzterem stammt auch das zweite Foto von Jørgensen. Das erste Foto ist aus der Wikipedia entnommen.






