Das ist harmlos. Ich hab so schon mal eine art "Lampe" für draußen gebastelt, sieht spät abends sehr cool aus. Technisch einziges Problem (abgesehen von der unangenehm riechenden und ungesunden aldehyd-emissionReosir hat geschrieben:Bei dem Video war ich echt froh, dass zwischen dem Experiment und mir einige Zeit und viele km Internet liegen! Wie die Spirale dann mal gewackelt hat, habe ich schon gedacht, die fällt jetzt rein und das ganze Teil zerreißt es
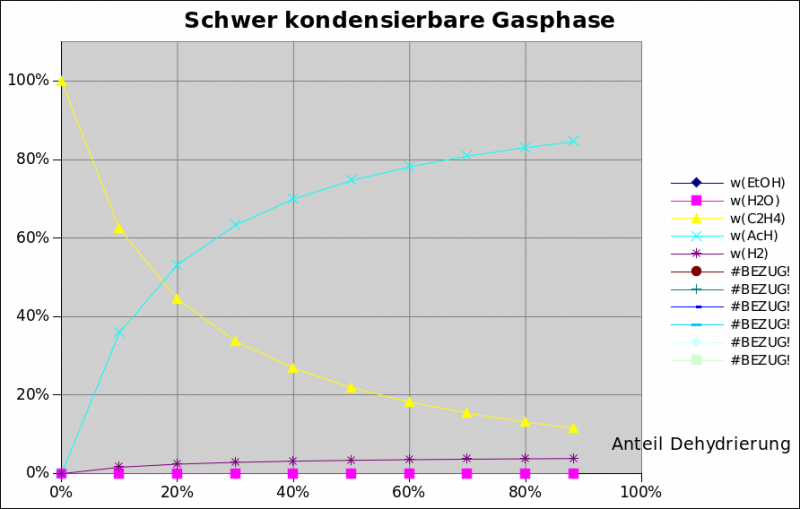
naja das mit einfach (Kupfer-)Draht in der Flüssigkeit heizen und Pyrolyse das ist dein Ansatz, den lass ich dich mal verfolgen, mal sehen was draus wird. Mit ist das zu unselektiv. Allzuviel Acetaldehyd entsteht vermutlich wohl eher nicht, wenn ich mir deine Gasvolumina ansehe sowie die Aussage "fruchtiger Geruch" (nennenswerte Mengen sind schon sehr beißend im Geruch, da ist Schluss mit fruchtig). Ich würde auch rein gefühlsmäßig die Dehydratisierung für die thermodynamisch bevorzugte Reaktion halten. Für Wasserstoffgas hab ich wenig Verwendung und davor hätte ich bzgl Explosionsrisiko wirklich Bammel. Eine Alkohol-Verpuffung ist schon unfein, Wasserstoff ist verheerend.Einige Probleme würden sich auflösen, wenn du die Dehydrierung ohne Sauerstoff machen würdest: kein Knallgas, keine große Abwärme. Und vielleicht könntest du den Wasserstoff noch für was Sinnvolles verwenden? Ich habe den Glühdraht ja auch absichtlich unter der Flüssigkeit gelassen, weil das Explosionen unwahrscheinlich macht. Acetaldehyd habe ich beim vorletzten Versuch mit Brennspiritus und Halogenlampe eindeutig riechen können. Wie wäre es einfach mal einen dünnen Kupferdraht (so 0,1 - 0,2 mm) elektrisch unter paar ml Ethanol zu heizen? Oder die Wendel einer Halogenlampe verkupfern?
Sonst habe ich im Zusammenhang mit dem Orsat noch die Dennis-Pipette und das Jäger-Röhrchen im Jander gesehen, die vom Funktionsprinzip her einen Ansatz liefern könnten.
Ich möchte jedenfalls die katalytische Oxidation benutzen wie sie auch in der Industrie zB zur Formaldehyd-Synthese eingesetzt wird - damit ist eine sehr hohe Spezifizität möglich und das Trennproblem beschränkt sich auf Alkohol vs Acetaldehyd. Ich dachte an so etwas wie einen aufsteigenden Rückflusskühler (Allihn) bei ca 20° Kühlwassertemperatur um den Alkohol zurückzuführen und einen absteigenden Intensivkühler bei ca 0° um Acetaldehyd zu kondensieren. Ist halt die Frage wie gut das rein praktisch trennt.
Als Reaktor dachte ich an einen Rundkolben mit einem warmen "Bodensatz" Ethanol in den ich durch ein Gaseinleitungsrohr dauernd Luft blubbere die evtl vorher schon durch Ethanol geleitet und vor-angereichert wird. Dadurch sollte die Mischung immer fett genug bleiben dass es zu keiner Ex-Atmosphäre kommt. OEG Ethanol ist mit 19 Vol-% ja relativ niedrig, das sollte den Prozess einigermaßen handhabungssicher machen. bei 40°C ist der Dampfdruck ca 200 mbar - also solange ich über der Temperatur bleibe müsste eine ausreichende Sättigung gewährleistet sein - solange ich nicht zu viel Luft rein blase - das ist der kritische Parameter! (Industriell führt man zB das Prozessgas im Kreis und zieht nur Formaldehyd über Wäscher heraus und speist so viel Methanol und Luft nach dass man mit dem O2-Gehalt immer so niedrig bleibt dass es zu keiner Ex-Atmosphäre kommen kann - N2 sammelt sich in dem System an. Bei meinem Ansatz wird sicher auch einiges an O2 verbraucht wodurch die OEG noch weiter sinkt). Ingesamt also vor allem die Frage wie kann ich die Luftzufuhr so dosieren dass mir die Reaktion weder abstirbt noch das Gemisch unter die OEG verdünnt wird.
Bleibt die Frage wie ich da drin am besten einen glühenden Kupferdraht-Katalysator mit ausreichend wirksamer Fläche eingefädelt und befestigt bekomme, und wie ich ihn auf Glühtemperatur vorgeheizt hinein bekomme. Und eben wie ich die Apparatur von außen gekühlt bekomme, denn wie gesagt die Abwärme ist beträchtlich und wenn man da nichts macht dann düfte spätstens nach einer Stunde das Glas nahe der Schmelztemperatur sein bzw aufgrund der Spannungen zum kalten Kühler einfach springen. Vielleicht indem man das ganze in ein zB 40-50° warmes Wasserbad stellt? Man will ja immer noch genug Ethanol verdampfen lassen um die Reaktion gut in Gang zu halten...
Also das ganze ist noch ein bisschen Nachdenk- und Tüftelarbeit, aber ansich nicht extrem problematisch (wenn man weiß was man tut