
Die NMR-Spektroskopie ist eine Sammlung analytischer Methoden, die das bei weitem wichtigste analytische Instrument der modernen organischen Chemie darstellt und auch enorme Bedeutung in der anorganischen Chemie, Biochemie und Biologie erlangt hat. Mit ihr lassen sich Molekülstrukturen aufklären, molekulare Konformationen bestimmen, intermolekulare Wechselwirkungen erforschen, die Identität und Reinheit von Substanzen ermitteln und vieles mehr.
Dieser Artikel soll einen kleinen Einblick in die Theorie und die Anwendungen diverser NMR-Verfahren geben, um ein gewisses Grundverständnis dieser Methoden zu vermitteln. Tiefergreifende Erklärungen sind schon aufgrund der enormen Fülle an verfügbaren Informationen nicht im Umfang dieses Artikels unterzubringen. Er beschränkt sich dabei auch auf die NMR-Spektroskopie an flüssigen Proben; zur für die Organische Chemie unbedeutenden Festkörper-NMR-Spektroskopie, aber auch für ein tieferes Verständnis der Theorie und Methoden sei auf die Literatur verwiesen. Zur Vereinfachung beschränkt der Artikel sich außerdem auf die Diskussion von Kernen mit einer Kernspinquantenzahl von I = ½, da diese Kerne (beispielsweise 1H, 13C, 19F, 31P) am wichtigsten sind.
Zunächst wird ein grober Überblick über die Theorie und die zugrundeliegenden Messmethoden gegeben, bevor die wichtigsten NMR-Experimente kurz beschrieben werden.
1. Das NMR-Experiment
Für Begriffserklärungen siehe Abschnitt 2.
Die Grundlage für sämtliche auf NMR basierenden Analyseverfahren ist die resonante Anregung von Atomkernen in einem starken Magnetfeld mit Radiowellen. NMR-aktive Kerne verhalten sich wie Stabmagneten und richten sich (genauer: ihren Kernspin) in einem statischen Magnetfeld B0 nach diesem aus. Diese Orientierung entlang des Magnetfelds ist energetisch günstiger, allerdings können sich die Spins auch entgegen dem B0-Feld orientieren, wenn ihnen die dazu nötige Energie zur Verfügung gestellt wird. Dies kann durch die Einstrahlung eines Breitband-Radiowellensignals um die Larmorfrequenzen der Probe erfolgen. In diesem Fall werden die Radiowellen absorbiert und die Kernspins orientieren sich gegen das B0-Feld, bevor sie wieder in ihre ursprüngliche Orientierung entlang B0 zurückkehren (relaxieren). Bei dieser Relaxation werden Radiowellen der Larmorfrequenzen emittiert, die gemessen werden (Abb. 1).

Abb. 1 – Die Orientierung und resonante Radiowellenabsorption/-emission von Kernspins bei NMR-Messungen.
Die so erhaltenen Daten sind noch kein fertiges Spektrum; man bezeichnet den Datensatz als FID (free induction decay, freier Induktionszerfall; Abb. 2), einen zeitlichen Abfall der Intensität der Radiowellenemission. Dieser kann aber am Computer durch ein mathematisches Verfahren, die sogenannte Fouriertransformation, zum entsprechenden NMR-Spektrum der Probe umgewandelt werden (Abb. 3).
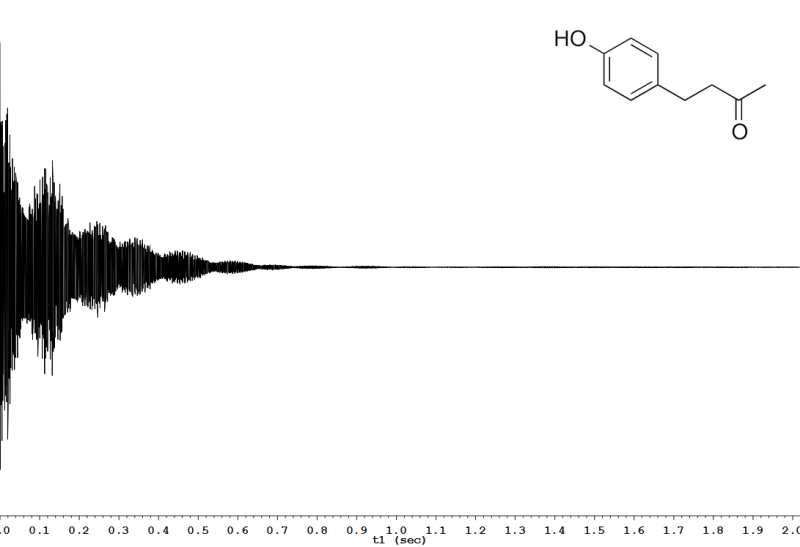
Abb. 2 – 600 MHz 1H-FID von Himbeerketon (4-(4-Hydroxyphenyl)-butan-2-on) in Chloroform-d.
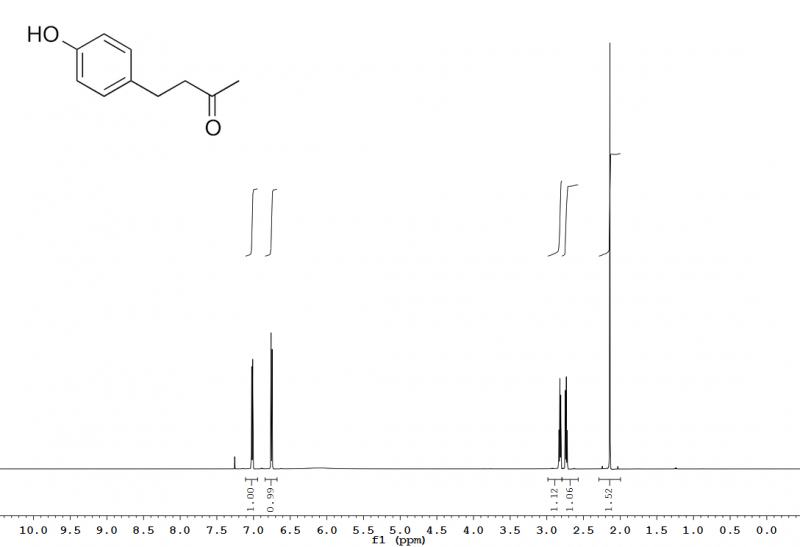
Abb. 3 – Aus dem FID (Abb. 2) erhaltenes 600 MHz 1H-NMR-Spektrum von Himbeerketon (siehe auch Abb. 10).
2. Begriffserklärungen
NMR – nuclear magnetic resonance (Kernspinresonanz), die resonante Absorption von Radiowellen durch Atomkerne mit einem magnetischen Moment, z. B. 1H, 2H, 13C, 19F, 31P u. v. a., die sich in einem starken externen Magnetfeld B0 befinden. Die auf dieser Resonanz basierende NMR-Spektroskopie erlaubt einen Einblick in die elektronische Umgebung einzelner Atome und ihrer Wechselwirkungen miteinander, somit also auch der Struktur komplexer Moleküle.
Kernspin – Gesamtdrehimpuls eines Atomkerns um seinen Schwerpunkt; dieser hängt mit einem magnetischen Moment zusammen, sodass Kerne als kleine Stabmagneten aufgefasst werden können und wie in dieser Modellvorstellung auch eine Ausrichtung des Kernspins entlang eines externen Magnetfelds erfolgt.
Larmorfrequenz – NMR-Resonanzfrequenz eines Nuklids, d. h. die Radiowellenfrequenz, die zur resonanten Absorption durch das Nuklid führt. Diese ist abhängig vom Nuklid und vom effektiven Magnetfeld Bef am gemessenen Kern, welches wiederum vom externen Magnetfeld B0 und, in geringerem Maße, von der elektronischen Umgebung am Kern abhängig ist. Beispielsweise absorbiert 1H in einem Magnetfeld von 14,1 Tesla Radiowellen bei 600 MHz; unterschiedliche elektronische Umgebungen, z. B. in CHCl3 gegenüber Si(CH3)4, bewirken Abweichungen von dieser Frequenz im Bereich einiger Hundert bis Tausend Hertz.
Chemische Verschiebung (δ) – die von der elektronischen Umgebung eines Kerns verursachte Abweichung seiner Larmorfrequenz von einem definierten Standard. Für 1H und 13C ist Tetramethylsilan Si(CH3)4 als Standard festgelegt; die entsprechenden Signale erscheinen in den 1H- bzw. 13C-NMR-Spektren bei jeweils 0,00 ppm. Die Angabe der chemischen Verschiebung in ppm hat den Vorteil, diese ansonsten magnetfeldabhängige Größe feldunabhängig zu machen. Beispielsweise entspricht eine chemische Verschiebung von 1,00 ppm in einem 300 MHz-NMR-Spektrum einer Verschiebung der Larmorfrequenz um 300 Hz, in einem 600 MHz-NMR-Spektrum um 600 Hz. Die chemischen Verschiebungen der Signale einer Probe in ppm sind somit für jedes Spektrometer, unabhängig von seiner Feldstärke, gleich. Elektronenziehende (elektronegative) Substituenten an einem Molekül führen zu einer sogenannten Tieffeldverschiebung (was aber einer Verschiebung zu höheren δ-Werten entspricht); umgekehrt führen elektronenschiebende Substituenten, insbesondere kovalent gebundene Metallatome, zu Hochfeldverschiebungen (zu niedrigeren δ-Werten).
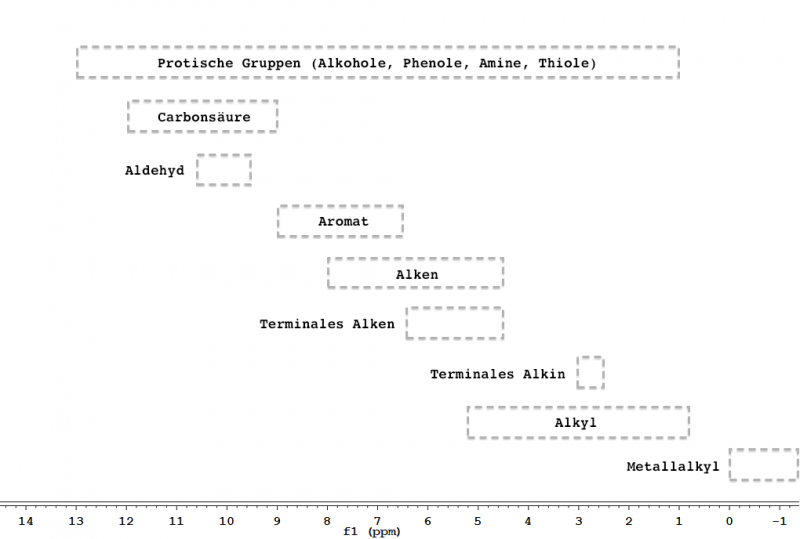
Abb. 4 – Typische chemische Verschiebungen in 1H-NMR-Spektren.
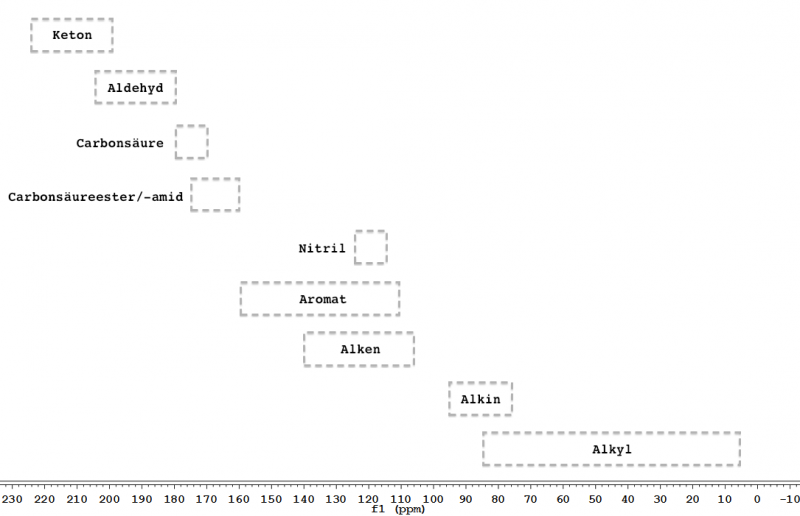
Abb. 5 – Typische chemische Verschiebungen in 13C-NMR-Spektren.
Polarisation – die Ausrichtung des magnetischen Kernspins nach einem magnetischen Feld. Mit steigender Ausrichtung der Spins der Probe erhöht sich auch die Stärke des gemessenen NMR-Signals und somit die Empfindlichkeit; da aber ein geringer Energieunterschied zwischen Kernen im ausgerichteten und im zufällig orientierten Zustand besteht, ist der Polarisationsgrad einer Probe im thermischen Gleichgewicht gering. Darauf beruht die vergleichsweise geringe Empfindlichkeit der NMR-Spektroskopie.
Chemische Äquivalenz – zwei oder mehr Kerne sind chemisch äquivalent, wenn sie sich in der selben elektronischen Umgebung befinden, wie beispielsweise die sechs 1H im Benzol oder die drei 1H der Methylgruppe in Toluol. Auch die beiden orthoständigen und die beiden metaständigen 1H im Toluol sind jeweils zueinander chemisch äquivalent (Abb. 6). Mehrere chemisch äquivalente Kerne geben ein einziges NMR-Signal (sind also im NMR-Spektrum anhand ihrer chemischen Verschiebung allein nicht unterscheidbar).
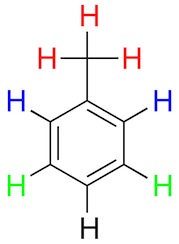
Abb. 6 – Das Toluolmolekül zur Veranschaulichung chemischer Äquivalenz. Chemisch äquivalente 1H sind jeweils in derselben Farbe dargestellt.
Magnetische Äquivalenz – zwei oder mehr Kerne sind magnetisch äquivalent, wenn sie neben chemischer Äquivalenz, also identischer chemischer Verschiebung, mit einem gemeinsamen Kopplungspartner auch dasselbe J-Kopplungsmuster ergeben. Magnetisch äquivalente Kerne sind somit immer chemisch äquivalent, aber chemisch äquivalente Kerne sind nicht zwangsläufig magnetisch äquivalent.
Skalare Kopplung (J-Kopplung) – die indirekte Wechselwirkung NMR-aktiver, magnetisch nicht-äquivalenter Kerne über chemische Bindungen (kovalente Bindungen, Wasserstoffbrückenbindungen). Diese bewirkt eine Aufspaltung der NMR-Signale, abhängig von der Anzahl beteiligter Kernspins und ist üblicherweise über bis zu vier kovalente Bindungen erkennbar (Abb. 11). Die Kopplung eines oder mehrerer (magnetisch äquivalenter) Kerne A mit einem weiteren, nicht magnetisch äquivalenten Kern B führt zur Aufspaltung des Signals von A in zwei gleich große Signale (ein sogenanntes Dublett). Der Abstand der beiden resultierenden Signale in Hertz ist magnetfeldunabhängig und wird als Kopplungskonstante J bezeichnet. Diese ist relativ charakteristisch und von den Kopplungspartnern, deren Abstand voneinander und deren Orientierung zueinander abhängig. Im obigen Beispiel zeigt auch das Signal von B eine Aufspaltung mit derselben Kopplungskonstante wie das Signal von A. So können über die Kopplungskonstanten strukturelle Beziehungen im Molekül hergestellt werden. Koppelt ein Kern C mit zwei benachbarten, zueinander magnetisch äquivalenten Kernen D, so ist das Signal für D wie oben beschrieben ein 1:1 Dublett, während das Signal für C zu einem 1:2:1 Triplett aufspaltet. Die sogenannte Multiplizität, also die Anzahl von Einzelpeaks, in die ein Signal aufspaltet, berechnet sich nach der Formel
M = n + 1
M = Multiplizität
n = Anzahl magnetisch äquivalenter Nachbarkerne
Die idealen Intensitätsverhältnisse der Einzelpeaks werden gegeben durch das Pascalsche Dreieck (Binomialverteilung). So hat ein Dublett ein Intensitätsverhältnis von 1:1, ein Triplett 1:2:1, ein Quartett 1:3:3:1, ein Quintett 1:4:6:4:1 und so weiter.
Dacheffekt – Abweichung der Intensitätsverhältnisse der Einzelpeaks eines Multipletts von der Binomialverteilung in einer solchen Weise, dass Peaks, die Kopplungen zueinander zeigen, ein "Dach" bilden (Abb. 7). Der Dacheffekt kann eine Hilfestellung bei der Zuordnung von Kopplungen liefern.
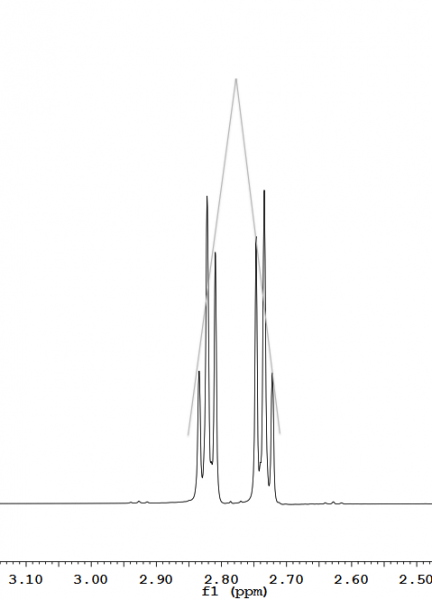
Abb. 7 – Dacheffekt zweier Peaks, die J-Kopplungen zueinander aufweisen.
Dipolare Kopplung – die direkte Wechselwirkung NMR-aktiver, magnetisch nicht-äquivalenter Kerne durch den Raum. Normalerweise werden diese Kopplungen in Lösung durch freie Beweglichkeit der Moleküle zeitlich gemittelt und sind somit nicht sichtbar, sind aber die Grundlage für den Kern-Overhauser-Effekt (nuclear Overhauser effect, NOE), der Übertragung von Polarisation durch den Raum zwischen nahe beieinanderliegenden Kernen. Dessen Messung im NOESY-Experiment kann wichtige strukturelle Informationen über Moleküle liefern.
Sensitivität – die Empfindlichkeit eines Kerns für eine NMR-Messung. Der empfindlichste stabile Kern ist 1H, gefolgt von 19F, welches 17 % weniger empfindlich als 1H ist, also bei einer Messung unter gleichen Bedingungen ein 17 % schwächeres Signal ergibt.
Rezeptivität – die Empfindlichkeit eines Nuklids für eine NMR-Messung. Neben der Sensitivität spielt hier auch die Häufigkeit des Nuklids im natürlichen Isotopengemisch des jeweiligen Elements eine Rolle. Die Rezeptivität ist das Produkt aus der Sensitivität und der natürlichen Häufigkeit des Nuklids. Bei Nukliden mit durch geringe natürliche Häufigkeit bedingter niedriger Rezeptivität, z. B. 13C oder 15N, kann es sinnvoll sein, die NMR-Messung an isotopenangereicherten Proben durchzuführen.
Heterokern – in der NMR-Spektroskopie wird jeder Nicht-1H-Kern, also z. B. 13C, 15N, 19F, 23Na, 31P, 59Co, 129Xe u. v. a., als Heterokern bezeichnet.
3. Praktische Einzelheiten zur Messung
Da Feststoffe im NMR-Spektrum ohne besondere Vorkehrungen äußerst breite, nicht interpretierbare Signale ergeben, müssen NMR-Messungen an klaren Lösungen der Proben durchgeführt werden. Die verwendeten Lösemittel sind meist deuteriert, um Störungen durch starke NMR-Signale des Lösemittels zu unterbinden und die Qualität der Spektren zu erhöhen. Für 1H-NMR-Spektren werden üblicherweise ca. 20 mg Probe in 0,7 mL eines geeigneten Lösemittels, meistens Chloroform-d, gelöst (bei Messungen von Nukliden mit geringer Rezeptivität empfiehlt sich eine höhere Probenkonzentration). Auch geringere Stoffmengen können gemessen werden; moderne Spektrometer können problemlos gute 1H-NMR-Spektren von Substanzmengen unter 1 mg aufnehmen. Das kostet jedoch deutlich mehr Messzeit. Die Lösung der Probe wird in ein Glasröhrchen von üblicherweise 5 mm Durchmesser (NMR-Röhrchen; Abb. 8) eingefüllt und dieses mit einer Halterung, einem sogenannten Spinner, in den Magneten des NMR-Spektrometers eingeführt. Die Aufnahme des Spektrums und die Auswertung der Daten erfolgt daraufhin am Computer. Die Aufnahme eines einzelnen Spektrums dauert üblicherweise wenige Sekunden; oft werden aber viele Spektren aufgenommen und addiert, um höhere Empfindlichkeiten (ein besseres Signal-zu-Rausch-Verhältnis) zu erhalten. Nach der Messung wird die Probe unverändert wieder entnommen; die NMR-Spektroskopie ist ein zerstörungsfreies Analyseverfahren.
Abb. 8 – Lösung einer Probe in Chloroform-d in einem NMR-Röhrchen.

Abb. 9 – Ein 600 MHz Varian VNMRS-600 NMR-Spektrometer (IOC, RWTH Aachen).
Die für NMR-Spektrometer angegebenen Frequenzen beziehen sich auf die 1H-Larmorfrequenz im jeweiligen Magneten. In Abb. 9 ist der Magnet die zylindrische, auf vibrationsdämpfenden Füßen stehende Konstruktion; er besteht aus einer mit flüssigem Helium gekühlten, supraleitenden Spule in einem Dewargefäß und erreicht eine magnetische Flussdichte von 14,1 Tesla (600 MHz 1H-Larmorfrequenz). Die Probe wird am gelblichen Anschluss oben mittig eingeführt, hier durch einen links davon befindlichen (matt-silbernen) Roboterarm. Die eigentliche Messung erfolgt durch den Probenkopf, der unten mittig eingeführt wird und die Sende-/Empfangsspule für die Radiowellen enthält. Vor dem Magneten ist ein ausgebauter Kryoprobenkopf zu sehen (senkrecht stehender, matt-silberner Stab mit roter Spitze), der zur Erhöhung der Messempfindlichkeit über die Schläuche mit Helium auf 20 K gekühlt wird. Der verwendete Radiowellenimpuls wird von einer NMR-Konsole (nicht im Bild) erzeugt, welche auch, nach Verstärkung des gemessenen NMR-Signals durch die Verstärker unten rechts neben dem Magneten, die weitere Verarbeitung des Signals zu einem FID übernimmt. Dieser wird dann an einem PC in ein NMR-Spektrum umgewandelt und weiter ausgewertet.
4. Messverfahren
Es gibt eine Reihe verschiedener NMR-Experimente verschiedener Dimensionalität. Jede Dimension stellt bei der graphischen Auftragung eine Frequenzachse dar. Am häufigsten werden normale, eindimensionale NMR-Spektren aufgenommen, die allesamt die Larmorfrequenzen der gemessenen Kerne auf der Frequenzachse (x-Achse) zeigen; die y-Achse gibt die einheitenlose Intensität des Signals. Zuordnungen von Signalen dieser Spektren entweder untereinander zwischen gleichen Kernen (beispielsweise die Bestimmung skalarer Kopplungen zwischen 1H in so genannten 1H-1H-COSY-Spektren) oder auch zwischen verschiedenen Kernen (beispielsweise die Bestimmung der Konnektivitäten zwischen 1H und 13C in so genannten 1H-13C-HSQC-Spektren) ist durch die Aufnahme zweidimensionaler Spektren möglich. Deren Messung ist sehr kompliziert; durch Aufnahme einer Reihe von 1D-Spektren mit inkrementeller Variation gewisser Messparameter können am Computer 2D-Spektren errechnet werden. Diese Spektren sind, wie der Name nahelegt, eine zweidimensionale Fläche mit x- und y-Achsen als Frequenzachsen; die Intensität ist hier in der dritten, meist nicht dargestellten Dimension. Peaks, die in diesen Spektren zwischen einem Peak des 1D-Spektrums der x-Achse und einem Peak des 1D-Spektrums der y-Achse auftreten, korrelieren diese Peaks miteinander. Bei 1H-1H-COSY-Spektren heißt dies beispielsweise, dass die entsprechenden 1H miteinander skalare Kopplungen aufweisen; bei 1H-13C-HSQC-Spektren heißt das, dass das jeweilige 1H mit dem entsprechenden 13C direkt kovalent verbunden ist (siehe Abb. 13 und 14). Es existieren auch mehrdimensionale (3D, 4D,...) NMR-Experimente; diese sind jedoch weniger wichtig und werden hier nicht behandelt.
4.1 1D-Experimente
1H-NMR-Spektroskopie
Dieses neben Relaxationsmessungen wohl einfachste NMR-Experiment erlaubt die Anzahl chemisch nicht-äquivalenter Wasserstoffatome (über die Anzahl von Peaks im gemessenen Spektrum), deren Art (aliphatisch, aromatisch,...; über die chemische Verschiebung), deren relative Mengenverhältnisse in der Probe (über die Integrale der Peaks) und in gewissem Umfang auch deren Position im Probemolekül (über skalare Kopplungen) relativ zueinander zu bestimmen (Abb. 10).
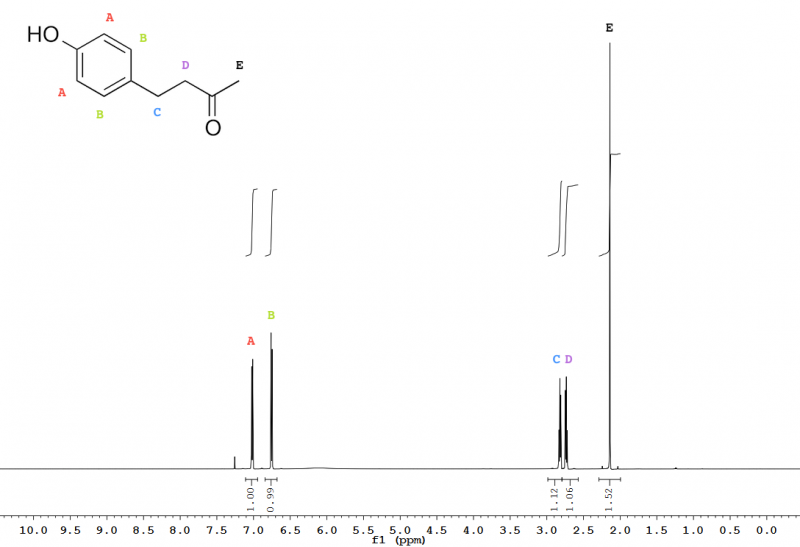
Abb. 10 – 600 MHz 1H-NMR-Spektrum von Himbeerketon (4-(4-Hydroxyphenyl)-butan-2-on) in Chloroform-d, mit Zuordnung (siehe auch Abb. 3). Die Hydroxylgruppe ist nur als geringfügige Erhebung der Baseline um 6 ppm zu erkennen; dies ist für protische Gruppen in Chloroform-d häufig.
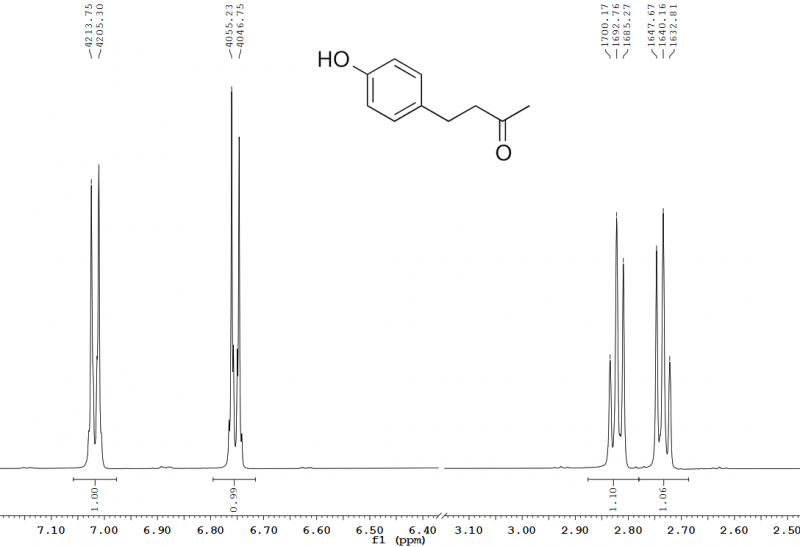
Abb. 11 – Ausschnitt aus dem 600 MHz 1H-NMR-Spektrum von Himbeerketon in Chloroform-d. Über den Peaks sind deren Frequenzen in Hertz angegeben, woraus sich die entsprechenden Kopplungskonstanten berechnen lassen. Die Kopplungskonstante für die aromatischen CH-Gruppen bei 6,75 ppm und 7,02 ppm beträgt 3JHH = 8,5 Hz, die für die CH2-Gruppen bei 2,73 ppm und 2,82 ppm beträgt 3JHH = 7,4 Hz. Bei 3JHH gibt die hochgestellte Drei die Anzahl der kovalenten Bindungen an, über die die Kopplung stattfindet, die tiefgestellten HH geben an, dass die Kopplung zwischen zwei 1H stattfindet.
13C-NMR-Spektroskopie
Dieses Verfahren ähnelt der 1H-NMR-Spektroskopie, allerdings ist die Rezeptivität 5700-fach geringer. Das liegt an der geringeren natürlichen Häufigkeit des 13C (1,07 % gegenüber 99,99 % für 1H) und der geringeren Sensitivität (0,016 relativ zu 1H). Da die Spektren zur Vereinfachung und zur Erhöhung der Messempfindlichkeit meist 1H-breitbandentkoppelt werden (also keine skalaren Kopplungen zwischen 13C und den daran gebundenen 1H auftreten) und die natürliche Häufigkeit von 13C so niedrig ist, dass nur sehr selten mehrere 13C benachbart sind, zeigen 13C-NMR-Spektren meist nur Singulett-Peaks (Abb. 12). Anders als bei der 1H-NMR-Spektroskopie sind 13C-NMR-Spektren nur unter sehr speziellen Messbedingungen quantitativ. Quantitative 13C-NMR-Spektren sind so messzeitaufwändig, dass sie nur sehr selten aufgenommen werden.
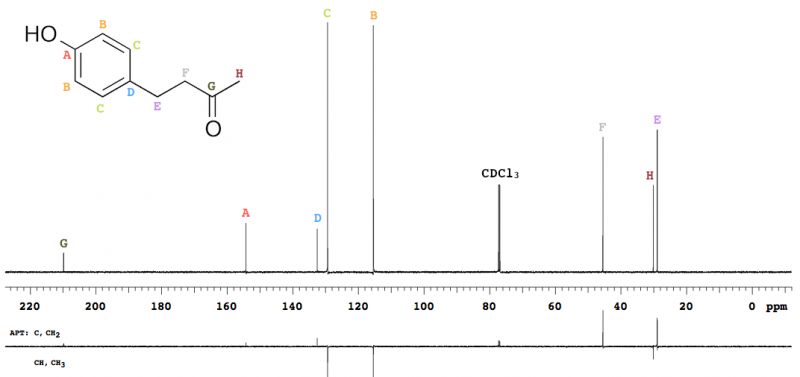
Abb. 12 – 151 MHz 13C-NMR-Spektrum von Himbeerketon in Chloroform-d, mit APT (siehe unten).
APT (Attached Proton Test)
Dieses Experiment wird oft als Ergänzung zu 13C-NMR-Spektren aufgenommen, da es eine Einteilung der Signale nach Anzahl der am zugrundeliegenden 13C gebundenen 1H ermöglicht. Die Messung ergibt ein 13C-NMR-Spektrum, bei dem allerdings solche 13C mit einer geraden Anzahl 1H (C, CH2) ein positives Signal ergeben, während solche mit einer ungeraden Zahl 1H (CH, CH3) in einem negativen Signal resultieren, was bei der Peakzuordnung hilft (Abb. 12).
DEPT (Distortionless Enhancement by Polarisation Transfer)
Diese Methode ist dem APT in ihrer Funktion ähnlich, kann aber mit höherem Messaufwand genauere Aussagen treffen. Es gibt verschiedene DEPT-Experimente, aus denen jeweils andere Informationen hervorgehen. DEPT 90 zeigt nur 13C-NMR-Signale von CH-Gruppen, während DEPT 135 positive Signale für CH und CH3 sowie negative Signale für CH2 ergibt. Im normalen DEPT-Experiment sind quartäre C-Atome nicht erkennbar, es gibt aber eine Variante des Experiments, bei der sie erhalten bleiben (DEPTQ).
Weitere Heterokern-NMR-Spektroskopien
Wichtig sind weiterhin insbesondere die 19F-NMR-Spektroskopie (natürliche Häufigkeit 19F: 100 %, Rezeptivität relativ zu 1H: 0,83, quantitative Messungen) und die 31P-NMR-Spektroskopie (natürliche Häufigkeit 31P: 100 %, Rezeptivität relativ zu 1H: 0,0066, normalerweise nicht-quantitative Messungen).
VT-NMR-Spektroskopie
Bei diesem Experiment werden normale eindimensionale NMR-Spektren (meist 1H) aufgenommen, allerdings bei Temperaturen unterhalb (bis ca. -80 °C) oder oberhalb (bis ca. 130 °C) der üblichen Raumtemperaturmessung. So können beispielsweise Tautomerie oder Konformationsänderungen, die über thermische Überwindung der Rotationsbarriere chemischer Bindungen ermöglicht werden, analysiert werden. Weiterhin ist durch Aufnahme einer Reihe von Spektren bei unterschiedlichen Temperaturen die Bestimmung der Enthalpien und Entropien solcher Umwandlungen möglich.
4.2 2D-Experimente
COSY (Correlation Spectroscopy)
Dieses Verfahren vereinfacht die Aufklärung skalarer Kopplungen in einem Spektrum, insbesondere dort, wo Peaküberlappungen die Bestimmung von Kopplungskonstanten unmöglich machen. Kreuzpeaks zeigen das Auftreten skalarer Kopplungen zwischen den zugrundeliegenden Kernen an (Abb. 13).
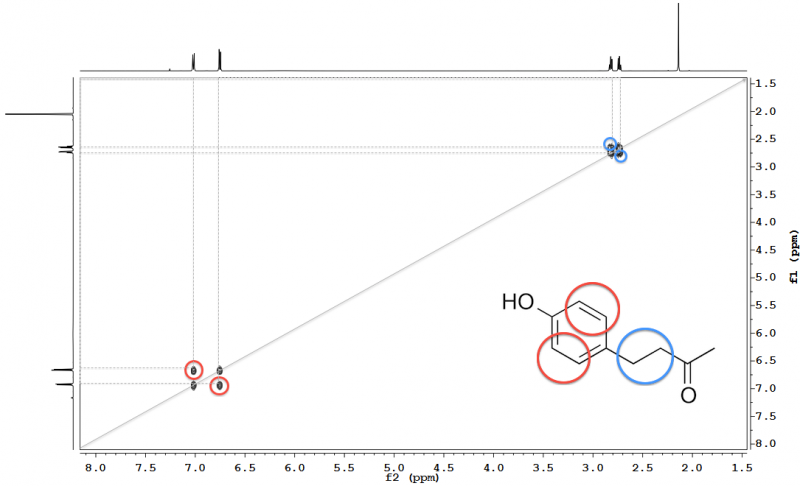
Abb. 13 – 600 MHz 1H-1H-COSY-Spektrum von Himbeerketon in Chloroform-d. Kreuzpeaks, d. h. solche, die die Signale chemisch nicht-äquivalenter 1H miteinander korrelieren, und die zugehörigen miteinander koppelnden Gruppen sind eingekreist; die Diagonalpeaks (d. h. die auf der das Spektrum teilenden Diagonalen) entsprechen einer Projektion des normalen 1D-1H-NMR-Spektrums und geben keine zusätzliche Information. Graue, gestrichelte Linien zeigen die Zuordnung der Peaks zu den entsprechenden Signalen der 1D-Spektren an.
NOESY (Nuclear Overhauser Effect Spectroscopy)
Dieses Experiment zeigt im Gegensatz zur COSY nicht Wechselwirkungen zwischen über Bindungen nahe beieinanderliegenden, sondern zwischen räumlich nahe gelegenen Kernen (normal bis 3,5 Å, maximal 5 Å Abstand). Kreuzpeaks zeigen räumliche Nähe der zugrundeliegenden Kerne an. Dies ermöglicht beispielsweise Konformationsbestimmungen.
HSQC (Heteronuclear Single Quantum Coherence)
Diese Methode ermittelt Konnektivitäten zwischen 1H und Heterokernen, meistens 13C. Peaks zeigen, welches 1H mit welchem Heterokern direkt, d. h. über nur eine einzige kovalente Bindung, verbunden ist (Abb. 14).
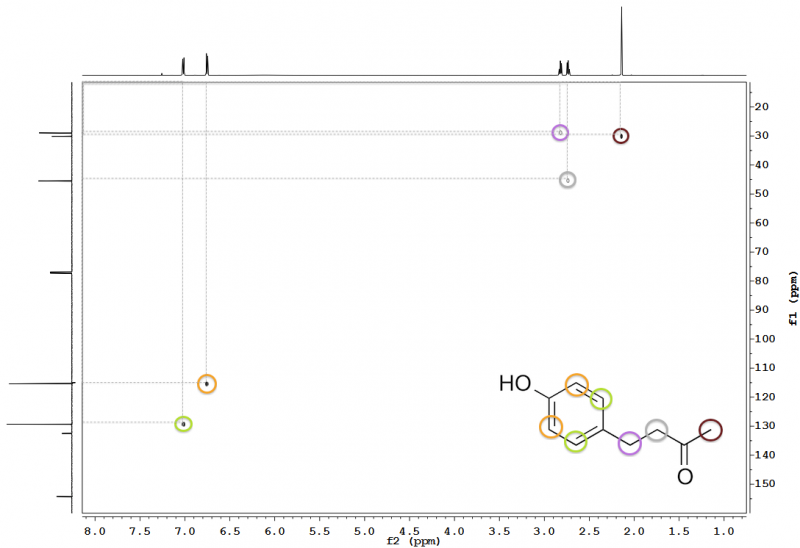
Abb. 14 – 600 MHz 1H-13C-HSQC-Spektrum von Himbeerketon in Chloroform-d. Peaks und die zugehörigen miteinander koppelnden CHn-Gruppen sind eingekreist. Graue, gestrichelte Linien zeigen die Zuordnung der Peaks zu den entsprechenden Signalen der 1D-Spektren an.
HMBC (Heteronuclear Multiple Bond Correlation)
Dieses Verfahren ermittelt ebenfalls Konnektivitäten zwischen 1H und Heterokernen wie 13C, wobei aber nur solche über mehrere Bindungen berücksichtigt werden. Peaks zeigen, welches 1H mit welchem Heterokern über zwei oder drei kovalente Bindungen verbunden ist. HSQC-artige Peaks über nur eine Bindung treten nicht auf.
DOSY (Diffusion-ordered Spectroscopy)
Dieses Pseudo-2D-Experiment teilt die Signale in der Frequenzdomäne nach den Diffusionskoeffizienten der zugrundeliegenden Moleküle auf, sodass eine Zuordnung der Signale in einem Stoffgemisch zu den unterschiedlichen enthaltenen Verbindungen (alle Signale einer Verbindung zeigen denselben Diffusionskoeffizienten) und eine Abschätzung der relativen Größe der jeweiligen Moleküle möglich ist. Aufgrund dieser Größensortierung und der „virtuellen“ Trennung des Stoffgemischs wird dieses Verfahren manchmal auch als NMR-Chromatographie bezeichnet. Die Integration der Diffusionsdomäne erlaubt quantitative Aussagen über die enthaltenen Stoffmengen der jeweiligen Substanzen, ähnlich der Integration eines Chromatogramms.
5. Zusammenfassung
Die NMR-Spektroskopie bietet eine reichhaltige Auswahl analytischer Verfahren für diverse Aufgabenstellungen, von der Strukturaufklärung kleiner Moleküle bis hin zu Proteinen, über Identitäts- und Reinheitsbestimmungen, Konformationsbestimmungen, die Bestimmung thermodynamischer Parameter von Reaktionen bis hin zur Erforschung intermolekularer Wechselwirkungen. Ihre enorme Bedeutung für die Naturwissenschaften allgemein wird ersichtlich an der Vergabe von sechs Nobelpreisen (an acht Wissenschaftler) in Physik, Chemie und Medizin für die Entwicklung von NMR-Theorie und -Verfahren seit 1943. In ihrer Aussagekraft ist sie unter den analytischen Verfahren in der organischen Chemie unübertroffen. Obwohl die vergleichsweise geringe Empfindlichkeit der Methode gegenüber beispielsweise Infrarot- und UV-Vis-Spektroskopie oder Massenspektrometrie insbesondere in der Anfangszeit der Anwendung der NMR-Spektroskopie in der organischen Chemie (1950/60er Jahre) gewisse Probleme darstellte, haben technische Fortschritte wie die Verwendung leistungsstarker, supraleitender Magnete und Kryomesstechnik sowie ausgefeilterer NMR-Methoden große Fortschritte gebracht. Problematisch sind noch die hohen Anschaffungskosten in Höhe mehrerer Hunderttausend bis Millionen Euro sowie hohe laufende Kosten für Kühlmittel und Reparaturen. Dafür steht mit der NMR-Spektroskopie aber ein Analyseverfahren zur Verfügung, das in einer Reihe naturwissenschaftlicher Disziplinen unentbehrlich geworden ist.
6. Literatur
Besonders empfehlenswert für den Anfang sind
- Friebolin, H. Ein- und zweidimensionale NMR-Spektroskopie: Eine Einführung. Wiley-VCH, Weinheim 2006.
- Berger, S., Sicker, D. Classics in Spectroscopy: Isolation and Structure Elucidation of Natural Products. Wiley-VCH, Weinheim 2009.
Danksagung
Danke an NI2 für hilfreiche Diskussionen und die Bereitstellung des Himbeerketons und an Prof. Dr. Markus Albrecht und Dr. Christoph Räuber der RWTH Aachen für die Aufnahme der NMR-Spektren.


