Ich habe noch ein Problem. Vielleicht weiß ja einer von euch weiter...?
Es geht um eine Arzneimittelanalyse. Dabei habe ich Extrakte des pulverisierten Arzneimittels mit wässrigen Lösungsmitteln hergestellt und dann bei verschiedenen pH mit Ether ausgeschüttelt. In den DCs der verschiedenen Fraktionen fallen drei Komponenten auf, von denen ich zwei identifiziert habe. Aber eine macht mir erhebliche Probleme. Sie tritt in verschiedenen anscheinend sehr unterschiedlichen Fraktionen auf und hat immer einen hohen Rf-Wert - in verschiedenen Laufmitteln.
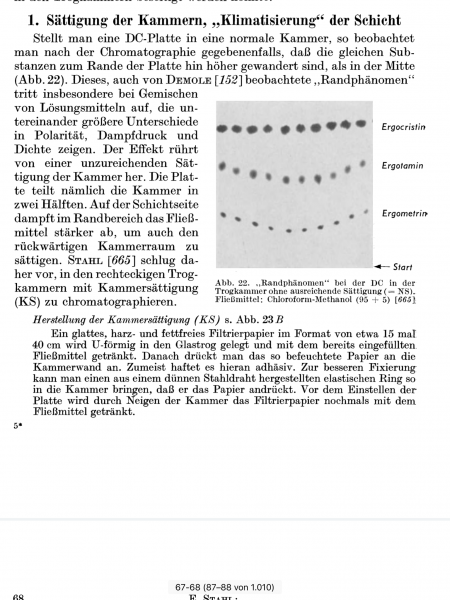
Folie 1: Extraktion mit NaHCO
3-Lösung, dann mit H
2SO
4 angesäuert, ausgeschüttelt, organische (Ether)Phase erneut mit verd. NaOH ausgeschüttelt und die verbleibende organische Phase (Fraktion A) chromatographiert. LM: Toulol+Methanol= 8+2.
(v.l.n.r.: Referenz 1, Referenz 1 und Fraktion A, Fraktion A, Referenz 2)
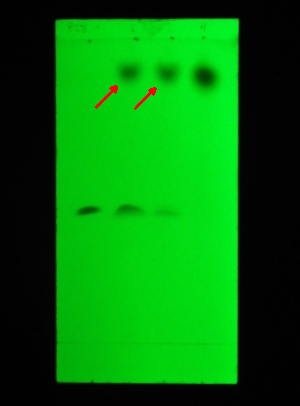
Folie 2: Extraktion mit verdünnter H
2SO
4 (0,5 N), dann neutralisiert und mit Weinsäure schwach angesäuert, ausgeschüttelt und organische Phase (Fraktion B) chromatographiert. LM: Ethylacetat+Methanol+Ammoniaklösung = 8,5 + 1,0 + 0,5
(v.l.n.r.: Referenz 3, Referenz 1, Fraktion B, Referenz 4 [gibt keine Fluoreszenzlöschung], Referenz 2)
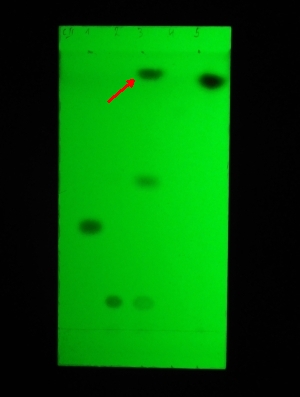
Folie 3: Die nach der zuletzt genannten Extraktion erhaltene wässrige Phase wird stark alkalisch gemacht (mit NaOH) und erneut mit Ether ausgeschüttelt. Die organische Phase ist Fraktion C. Danach wird die wässrige Pase neutralisiert, mit Ammoniak schwach alkalisch gemacht und nochmals ausgeschüttelt (Fraktion D). Die organischen Pasen werden chromatographiert. LM: gleiches wie unter 2
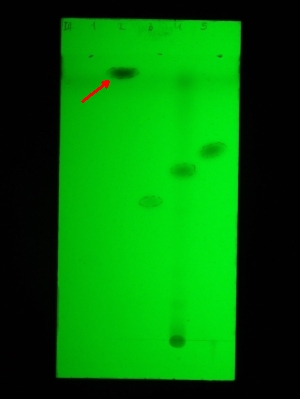
(v.l.n.r.: Referenz 5 [gibt keine Fluoreszenzlöschung], Fraktion C, Referenz 6, Fraktion D, Referenz 7)
Referenz 2 (in Folie 1 und 2 die ganz rechte Spur) ist Sudan III und in allen Chromatografiebeispielen im Auterhoff-Kovar stets die Substanz mit dem höchsten RF-Wert. Die gesuchte Substanz hat einen höheren Rf (was im Buch nicht vorkommt) muss also vermutlich ungewöhnlich lipophil sein. Sie lässt sich sowohl mit schwach alkalischer als auch stark saurer Lösung extrahieren und aus stark saurer, schwach saurer und stark alkalischer Lösung ausschütteln. In Fraktion D ist sie nicht mehr vorhanden - wohl weil sie bei den beiden vorigen Extraktionen (Fraktion B und C) vollständig entfernt wurden.
Diese vermuteten Eigenschaften passen mir aber nicht recht zusammen: lipohil, wasserlöslich, sowohl aus saurer wie auch aus alkalischer Lösung gut extrahierbar?
Ich erwarte nicht, dass ihr mir sagt, um welche Substanz es sich handeln könnte, aber vielleicht eine Stellungnahme zu folgenden Fragen:
Stimmen meine Anahmen über das Verhalten der Substanz?
Sind die beiden Laufmittel in ihren Eigenschaften möglicherweise sehr ähnlich (und deswegen beidesmal ein so hoher Rf)
Hat jemand einen Kommentar? Sonst eine Idee?
"Alles sollte so einfach wie möglich gemacht werden. Aber nicht einfacher." (A. Einstein 1871 - 1955)
"Wer nur Chemie versteht, versteht auch die nicht recht!" (G.C. Lichtenberg, 1742 - 1799)
"Die gefährlichste Weltanschauung ist die Weltanschauung der Leute, die die Welt nie gesehen haben." (Alexander v. Humboldt, 1769 - 1859)