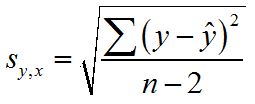Offenbar sind die Regressionsgeraden doch verschieden, je nachdem ob man alle Originalwerte einsetzt oder ob man zunächst die differierenden, bei der gleichen Konzentration gemessenen Werte mittelt und die Mittelwerte einsetzt.Pok hat geschrieben:Ich hab die Werte jetzt auch mal grafisch dargestellt, bekomme aber eine leicht andere Gleichung:
y = 15,40229 x - 0,07384
R² = 0,99929
Das kommt jedenfalls raus, wenn man die Mittelwerte jeder Eich-Messung (inklusive Nullwert) nimmt. Wenn man dann die Mittelwerte der Proben eingibt, kommt man auf 83,3 und 165,5 mg/l. Mit der 10-fachen Gewichtung von (0|0) kommt man auf 84,5 und 165,7 mg/l. Der Unterschied ist wirklich gering und schon die Ungenauigkeit bei der einen Messung (0,272 zu 0,275) entspricht eine größeren Abweichung.
Bei Komplexen kann ich das nachvollziehen.Xyrofl hat geschrieben:Man könnte ja vermuten, dass der lineare Bereich für ganz kleine Konzentrationen verlassen wird und somit eine Extrapolation auf (0|0) nicht korrekt sein muss, wie in deinem Beispiel mit den Komplexen. Lineare Regression mit freien Y-Achsenabschnitt ist meiner Erfahrung nach auch das übliche Verfahren, vermutlich nicht ohne Grund.
Aber bei der Absorption von Licht durch eine gefärbte Substanz gilt das Lambert-Beer'sche Gesetz soweit ich weiß bis an den Nullpunkt, und wo nichts ist kann ja auch keine Absorption eintreten. Also würde ich in diesem Fall dafür plädieren, daß die Gerade durch den Ursprung gehen muss. Der Ursprung ist sozusagen der einzige Punkt, der nicht fehlerbehaftet sein kann.
Angenommen bei meiner Eichmessung streuen die ersten beiden Punkte - nahe dem Ursprung - etwas nach oben und die beiden letzten etwas nach unten. Dann kann ich eine Gerade anfitten, die vielleicht ideal durch alle Punkte geht (mit einem positiven Y-achsenabschnitt) - aber bei der nächsten Messung streut ein Wert nahe dem Ursprung etwas nach unten, statt nach oben. Dann ist der Fehler größer als wenn ich die Eichgerade durch den Ursprung lege. In diesem Falle weichen alle Eichpunkte etwas von der Geraden ab: bei niedrigen Konzentrationen läuft sie unterhalb, bei höheren etwas oberhalb der Messwerte. Vorausgesetzt ist dabei natürlich die Annahme, dass die Abweichung wirklich nur durch einen stochastischen, und nicht durch einen systematischen Fehler bedingt ist.
@Pok: kann das Calc-Programm von Open Office das?Xyrofl hat geschrieben:Den Standardfehler der Regression kann man wie die Standardabweichung einer Verteilung berechnen, nur eben statt der Abweichung vom Mittelwert die Abweichungen von der Regressionsfunktion verwenden.
Das sollte man aber nicht selbst machen müssen, eigentlich sollten die entsprechenden Statistikprogramme das können.