Die Geschichte des Murexids ist bereits so alt wie die Geschichte der modernen Chemie, führt uns fast 250 Jahre zurück und hat viele berühmte Namen beschäftigt.
Bereits 1776 entdeckte Carl Willhelm Scheele[1] es bei seinen Untersuchungen von Harnsteinen: als er sie mit Salpetersäure auflöste und Ammoniak zusetzte, erhielt er eine kräftig violette Farbe und hatte im Prinzip den "Murexid-Test" erfunden. William Hyde Wollaston und William Prout (berühmt geworden durch die "Prout'sche Hypothese nach der die Atommasse jedes Elements ganzzahliges Vielfaches der Atommasse des Wasserstoffs sei) ordnete sie 1818 erstmals richtig dem Ammoniumsalz der Purpursäure zu und gab ihr auch den Namen.
Liebig und Wöhler[3] beschäftigten sich in ihrer Arbeit "Untersuchungen über die Natur der Harnsäure" mit der Untersuchung der Reaktionsprodukte der Harnsäure - unter anderem auch mit Salpetersäure. Neutralisierten sie das Reaktionsprodukt in der Hitze, erhielten sie Ammoniumpurpurat. Sie führten dafür auch den Namen Murexid ("murex-" in Anlehnung an Purpur, der lateinische Gattungsname der Purpurschnecke ist Murex; "-id" in Anlehnung an die vermutete Amidstruktur) ein und zeigten einen weiteren, zuverlässigeren Syntheseweg durch Oxidation aus Uramil (5-Aminobarbitursäure) mit Quecksilberoxyd. Sie waren von dieser Verbindung fasziniert, so schrieben sie:
Die ermittelte Summenformel C12H12N10O8 war zwar falsch und auch die Einschätzung, dass es sich um ein Amid und kein "Ammoniaksalz im gewöhnlichen Sinne" handelt, nur teilweise richtig. Richtig erkannten sie jedoch, dass die freie Purpursäure nicht existiert, sondern dass Murexid beim Ansäuern unter anderem wieder in Uramil zerfällt. Recht behalten sollten Sie auch mit der einleitenden begeisterten Bemerkung über die Vielfalt der Verbindungen die sie aus Harnsäure synthetisieren konnten:...dies führte uns nun zu einer Herleitungsmethode dieses merkwürdigen Produkts, welches an Schönheit alles übertrifft was die Chemie darbietet...
Gregory[4] optimierte 1841 die Reaktionsbedingungen für die Synthese von Alloxan aus Harnsäure, aus dem man Alloxantin und in weiterer Folge Murexid gewinnen kann. Fritzsche korrigierte die Formel später zu C16H8N6O11, Gmelin in seinem Handbuch wiederum zu C16H8N6O12 und stellte es durch Überleiten von NH3 auf Alloxantin her. Beilstein[5] passte die Synthesevorschrift von Liebig und Wöhler aus Uramil an und bestätigte Gmelins - immer noch falsche - Formel. Weiters stellte er die Amid-Hypothese von Liebig und Wöhler in Frage, zeigte dass das von ihnen gefundene "Murexan" tatsächlich identisch mit Uramil ist und stellte einige andere Salze der Purpursäure her.Die Philosophie der Chemie wird aus dieser Arbeit den Schluß ziehen, daß die Erzeugung aller organischen Materien, insoweit sie nicht mehr dem Organismus angehören, in unseren Laboratorien nicht allein wahrscheinlicher, sondern als gewiß betrachtet werden muß. Zucker, Salicin, Morphin werden künstlich hervorgebracht werden. Wir kennen freilich die Wege noch nicht, auf dem dieses Endresultat zu erreichen ist, weil uns die Vorderglieder unbekannt sind, aus denen diese Materien sich entwickeln, allein wir werden sie kennen lernen.
Stenhouse[6] bemerkte 1843 dass auch Thein (Coffein) eine Murexidreaktion ergibt, Rochleder [7] beschäftigte sich 1849 mit den Oxidationsprodukten des Coffeins und stellte durch Oxidation mit Chlor Dimethylalloxan und Dimethylalloxantin her - letzterer Verbindung gab er den Namen "Amalinsäure". Auch hier konnte er eine Murexidreaktion bemerken, stellte aber fest:
Rochleder stellte auch die Hypothese auf, dass Coffein und Theobromin verwandt seien. Ludwig Medicus[8] schließlich zeichnete 1875 erstmals die richtigen Strukturen der Harnsäure und der Purinbasen Xanthin, Guanin, Coffein, Theobromin (hier lag er noch falsch mit der Position der zweiten Methylgruppe) sowie von Hypoxanthin."Der hierbei gebildete Körper löst sich in Wasser mit der Farbe des Murexids auf, man erhält aber aus dieser Lösung keine Krystalle von Murexid, ebenso wird die Flüssigkeit durch Zusatz von Kali sogleich entfärbt, ohne daß zuvor die Farbe in Blau übergeht."
Maly[9] erwähnte 1881 bereits "die nahe verwandtschaftliche Stellung, welche man den beiden Pflanzenbasen Caffein und Theobromin zur Harnsäure gewöhnlich anweist" und zeigte dass sich Coffein und Theobromin um eine Methylgruppe unterscheiden. Seine Summenformeln waren bereits korrekt. In einer weiteren Arbeit[10] 1882 prüfte er Rochleders Darstellung der Amalinsäure aus Coffein und Chlorat und korrigierte auch bereits auf die richtige Summen- und Strukturformel!
Emil Fischer[11] systematisierte 1882 die Darstellung der Amalinsäure weiter und untersuchte auch viele andere Coffein-Derivate. Falsch lag er jedoch bei den daraus gezogenen Schlüssen und der Strukturformel des Coffeins (er zweifelte Medicus' Strukturvorschlag an): ein Sauerstoff landete im falschen Ring - folglich war auch seine Ansicht über Xanthin und Theobromin nicht richtig. Die Murexidprobe benutzte auch er. Heinrich Biltz[12] schließlich optimierte die Darstellung der Amalinsäure erneut, indem er zur Reduktion Zinnchlorid einsetzte. Seine Vorschrift findet sich bis heute als Referenz in Papers die von dieser Vorläuferverbindug des Murexids (bzw. Tetramethylmurexids) ausgehen.
Wann genau die Struktur des Murexids und seiner Derivate feststand, konnte ich nicht ganz nachvollziehen - 1936 rätselte Davidson [13] wegen Unklarheiten bzgl des Mechanismus der Bildung und der sauren Hydrolyse immer noch über den (bereits richtigen) Vorschlag von Piloty, Möhlau, Slimmer und Stieglitz aus dem Jahr 1904. Davidson stellte murexid durch Umsetzung von Alloxantin in siedendem Eisessig dar[14].
Einzug in die Analytik hielt das Murexid schließlich mit der Arbeit von Schwarzenbach und Gysling[15] 1949 - sie benutzten Murexid als Indikator auf Calcium- und andere Metall-Ionen. In einer weiterführenden Arbeit[16] wurden schließlich auch die strukturanalogen Verbindungen synthetisiert und untersucht. Autherhoff[17] stellt weitere Murexid-Derivate unterschiedlicher Methylierung her und untersuchte sie. Und dieses Molekül ist nach wie vor nicht aus der Mode - In Google Scholar findet man in den ersten 4 Monaten 2020 bereits 96 Verweise auf "Murexid"!
Von diesen Verbindungen habe ich mir das Tetramethylmurexid ausgesucht, da es aus einfach und billig zugänglichem Coffein leicht nach dem Verfahren von Biltz[12] mit geringer Modifikation herstellbar ist und auch einige interessante analytisch-chemische Eigenschaften hat, die es vom "kommerziellen" Murexid unterscheiden.
Geräte:
500 ml Zweihals-Rundkolben, Magnetrührer, Wasserbad mit Thermostat, Tropftrichter, Gaseinleitungsrohr, Sinternutschen, Bechergläser
Chemikalien:
Coffein

Salzsäure

Natriumchlorat


Zinn(II)chlorid

Wasserstoffperoxid
Tetramethylalloxantin

Ammoniumhydrogencarbonat
Tetramethylmurexid
Hinweis:
Zum Zwischenprodukt Tetramethylalloxantin bzw. zum Produkt Tetramethylmurexid konnte ich keine gesicherten toxikologischen Einstufungen in der Literatur finden. Das Analog Alloxan ist jedoch dafür bekannt, durch Zerstörung der insulinproduzierenden Zellen Diabetes auszulösen, daher ist im Umgang damit höchste Vorsicht geboten!
Bei der Synthese entsteht Chlorgas bzw. kann Chlorcyan entstehen, es ist daher unbedingt in einem Abzug bzw. im Freien zu arbeiten!
Durchführung:
In einem 500 ml Zweihals-Rundkolben werden 25 g (0,129 mol) Coffein vorgelegt und unter Rühren in 35 ml konz. (37%) HCl (ca 0,42 mol) und 75 ml dest. Wasser gelöst. Um das Ausströmen von Chlorgas bzw. HCl-Dämpfen zu minimieren wird ein Rückflusskühler aufgesetzt, jedoch nicht mit Wasser betrieben. Auf einem Hals des Kolbens wird ein Gaseinleitungsrohr vorgesehen, das mit einem Schlauch mit einer kleinen Aquarienpumpe für Luft verbunden war, jedoch anfangs noch nicht eingeschaltet. Später wird es gegen einen Glasstopfen getauscht, um bequem Chlorat zugeben zu können.
Der Ansatz wird auf einem Wasserbad, das auf 50 °C eingestellt ist, temperiert und dann in kleinen Portionen über eine Zeit von 2-3 Stunden verteilt 8,7 g Natriumchlorat unter kräftigem Rühren zugegeben. Die langsame Zugabe ist dabei für einen Erfolg essentiell wichtig, da ein Überschuss zu einem vermehrten Abbau des Coffeins über das Zwischenprodukt Alloxan hinaus führt! Man kann es z.B. gut damit abschätzen, dass sich durch das gebildete Chlor eine gelbliche Farbe im Ansatz bzw. der Gasphase darüber erkennen lässt - erst wenn diese merklich nachgelassen hat sollte die nächste Portion (kleine Spatel voll) zugegeben werden. Bereits nach kurzer Zeit beginnt die Ausscheidung eines weißen Niederschlags aus 8-Chlorcoffein. Die Menge an Niederschlag nimmt immer mehr zu, und der Ansatz wird so dick, dass er nur noch schwer gerührt werden kann. Um die Rührbarkeit etwas zu unterstützen, werden nochmal ca. 20 ml halbkonz. Salzsäure zugegeben. Erst bei ca. 3/4 der Menge an Chlorat kommt es wieder zu einer merklichen Auflösung, eine vollständige Auflösung des Niederschlags erfolgt erst wenn alles Chlorat zugegeben ist.
Nach Beendigung der Zugabe wird noch ca. 30 min weiter gerührt, dann ca. 15 min am warmen Wasserbad Luft durch den Ansatz geblasen um überschüssiges Chlor zu entfernen, schließlich wird das Wasserbad gegen eine Eiskühlung getauscht und weitere 15 min Luft durchgeblasen. Der Rückflusskühler wird nun gegen einen Tropftrichter getauscht und eine Lösung von 14,1 g Zinn(II)Chlorid (als SnCl2 0,5 H2O; 0,071 mol) in 20 ml halbkonz. HCl in den kalten Ansatz langsam eintropfen gelassen. Bereits nach kurzer Zeit scheidet sich ein weißer, feinkristalliner Niederschlag ab, nach Beendigung der Zugabe wird nochmal ca. 30 min weiter gerührt.
Nun werden 5 ml 35%iges Wasserstoffperoxid zugegeben, worauf sich die Menge an Niederschlag nochmal merklich erhöht. Nach weiteren 30 min Rühren am Eisbad wird der Niederschlag abgenutscht, gut mit kaltem Wasser nachgewaschen und an der Luft an einem warmen Ort getrocknet.
Zur Reinheitskontrolle wurde eine DC gemacht - Tetramethylalloxantin löst sich in fast keinem Lösungsmittel, lediglich in DMF. Als Laufmittel diente Aceton:Chloroform:Wasser 15:4:1. Im Produkt wurden keine Verunreinigungen, insbesondere kein Coffein gefunden.
Ausbeute: 10,1 g Tetramethylalloxantin (45,8% d.Th.)
Zur Umsetzung zum Tetramethylmurexid werden 3,0 g Tetramethylalloxantin (8,8 mmol) im Mörser sehr fein gepulvert und in einem 25 ml Becherglas vorgelegt. Dazu werden 3,0 g Ammoniumhydrogencarbonat (38 mmol, ein mehr als doppelter molarer Überschuss) gegeben und das ganze rasch und so innig wie möglich vermischt. Sofort bei Kontakt der beiden Stoffe bildet sich eine violette Färbung die rasch intensiver wird und ins Bräunliche übergeht. Das Becherglas wird mit einem Uhrglas abgedeckt und der Ansatz über Nacht an einem warmen Ort (z.B. auf dem Heizkörper) stehen gelassen. Am nächsten Tag wird das Becherglas zunächst wieder abgedeckt und nach weiteren ca 12 Stunden im Trockenschrank bei 110 °C nochmal 2 Stunden getrocknet. Nach dem Erkalten kann die poröse Masse in einer Porzellanschale leicht zu einem intensiv ziegelroten Pulver zerrieben werden.
Zur Reinheitskontrolle wurde eine DC gemacht - Tetramethylmurexid löst sich in fast keinem Lösungsmittel, lediglich in Wasser oder DMF. Als Laufmittel diente Aceton:Chloroform:Wasser 15:4:1. Das Produkt neigt zur Zersetzung bzw. laufen ionische Verbindungen auf Kieselgel generell schlecht - im Produkt wurden jedoch keine Verunreinigungen durch Tetramethylalloxantin mehr gefunden. Schwarzenbach und Gysling[16] kristallisierten noch 2 x durch Auflösen in Wasser und Fällung durch Aussalzen mit Ammoniumchlorid um und erhielten so ein analytisch reines Produkt. Dies erschien hier aber nicht unbedingt erforderlich.
Ausbeute: 3,0 g Tetramethylmurexid (100% d.Th.)
Achtung: es ist hier wichtig, den Ansatz nach dem anfänglichen Vermischen nicht mehr erneut durchzurühren! Durch die Reaktion wird Wasser freigesetzt sodass der Ansatz beim Rühren zu einem klebrigen festen Klumpen zusammenbacken würde der nur schlecht trocknet bzw. nur schwer gepulvert werden kann. Sollte das passieren, so kann man die Masse in siedendem Alkohol suspendieren - Tetramethylmurexid löst sich darin nur schlecht, und so erhält man wieder ein feinpulvriges Produkt das abgenutscht werden kann.
Charakterisierung:
Murexid ist in der Analytischen Chemie beliebt als metallochromer Indikator zur Bestimmung von Ca, Cu, Ni oder Co. Tetramethylmurexid sollte dafür auch gut geeignet sein, da es nach wie vor das gleiche chelatbildende Strukturelement enthält:
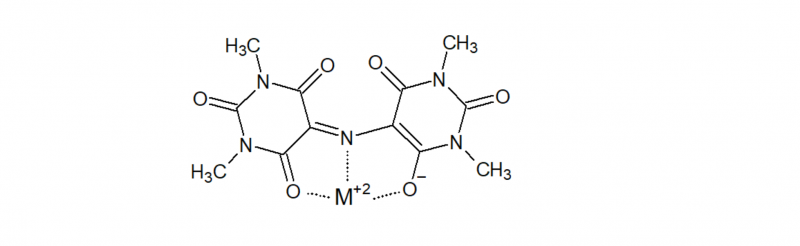
Der wesentliche Unterschied ist jedoch, dass alle NH durch CH3 substituiert sind, wodurch die Säure-Basen-Eigenschaften, die sich in der "5-basigen Säure" Murexid durch die Tautomerie:
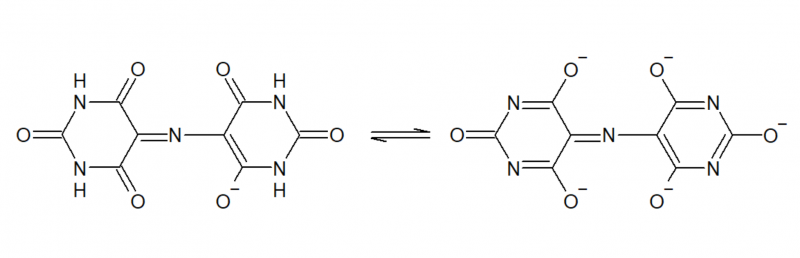
ergeben, wegfallen. Tatsächlich zeigt Tetramethylmurexid keine pH-abhängigen Farbumschläge, es zersetzt sich lediglich bei hohem (>10) oder niedrigem (<4) pH mehr oder weniger rasch durch Zerfall in seine Bestandteile. Der Vorteil ist daher, dass es im Gegensatz zu Murexid auch bei pH-Werten <8 gut einsetzbar ist bei denen Murexid sonst eine orange Eigenfarbe hat, wodurch der Umschlag bei einer Titration mit EDTA nicht mehr klar wahrnehmbar wäre. Das empfindliche Spiel mit der richtigen Dosierung von NH3 kann somit entfallen - denn eine zu große Menge an NH3 wäre auch wieder störend da sie durch Amin-Komplexbildung die Titration ebenfalls stört.
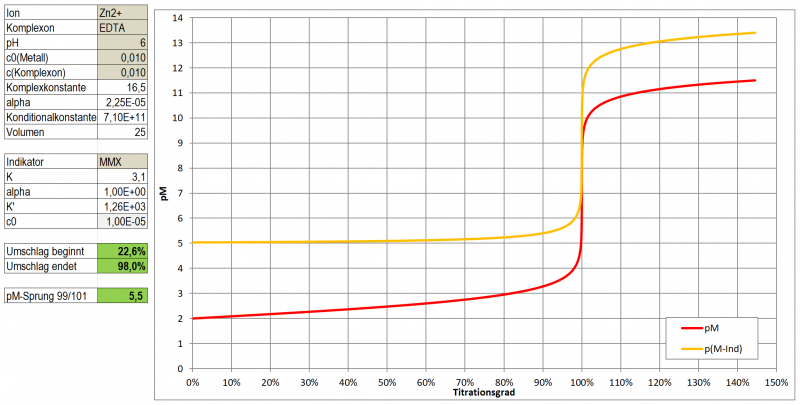
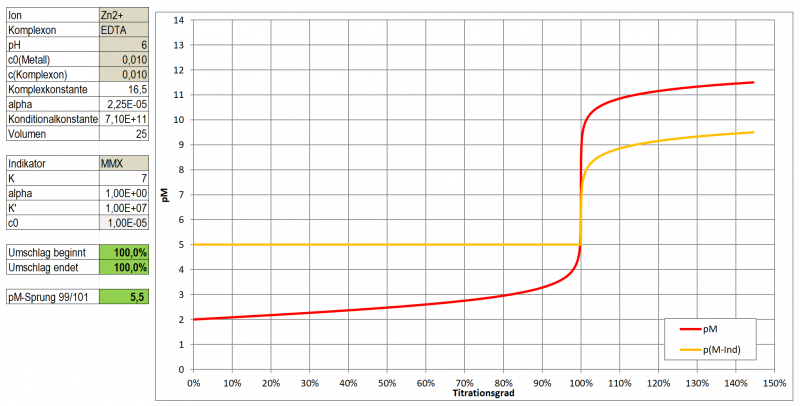
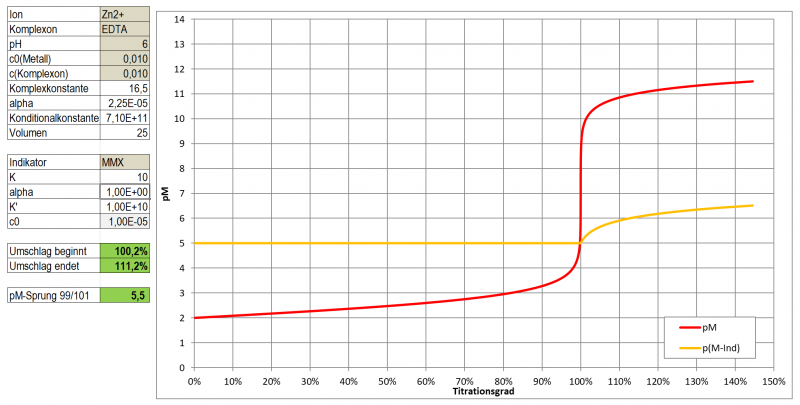
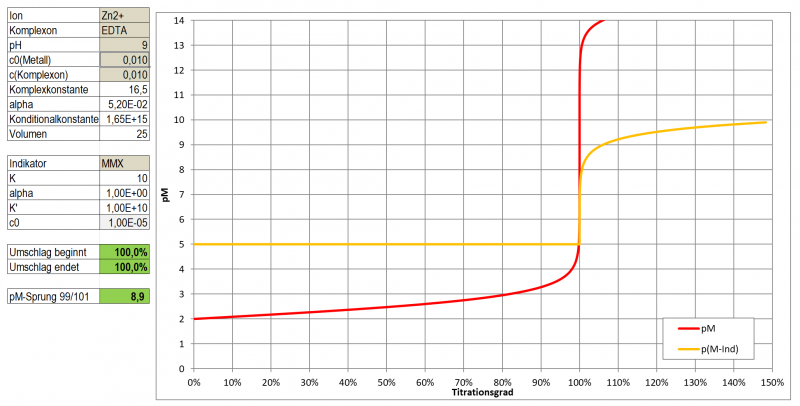
In einem ersten Test zeigt Tetramethylmurexid im Bereich pH 5 - 8 sehr kontrastreiche Farbumschläge zu Gelb-Orange-Tönen mit Cu, Ni, Co, Zn und Cd. Mit Pb ergibt sich eine wenig kontrastierende, kirschrote Färbung, Ca ergibt bei pH 10 eine wenig kontrastierende, dunkle Orange-Tönung.
Ein optimaler Indikator für die komplexometrische Titration ist es aber dennoch nicht. Wie ein Test mit Zn zeigte, ist der Farbumschlag doch eher schleifend (vermutlich einer etwas zu geringen Komplexbildungskonstante geschuldet). Hat man eine Vergleichslösung, dann kann man natürlich auf das reine Magenta des freien Indikators hin arbeiten und kann durchaus Ergebnisse mit <3 % Abweichung erhalten, die letzten Tropfen sind dabei aber schwer einzuschätzen. Möglicherweise könnte es aber gut als Farbreagens für die Photometrische Bestimmung der genannten Metalle im Spurenbereich (unter 10-4 mol/l) geeignet sein.
Die Charakterisierung mit dem Photometer zeigt ein ausgeprägtes Absorptionsmaximum bei 530 nm, der gelbe Komplex mit Zn hat das Maximum hingegen bei 461,5 nm.
Entsorgung:
Abfälle kommen zu den halogenhaltigen organischen Abfällen.
Erklärung:
Coffein ist ein Purinalkaloid aus der Stoffgruppe der Xanthine. Alle NH- Gruppen des Xanthins sind darin durch Methylengruppen substituiert, was seine sehr hohe chemische Stabilität bzw. geringe Reaktionsfreudigkeit erklärt. Lediglich die CH-Gruppe des Imidazolrings (Position 8 ) ist noch für einen Angriff frei - hier erfolgt im ersten Schritt eine Substitution durch Chlor das sich aus Chlorat und HCl bildet:
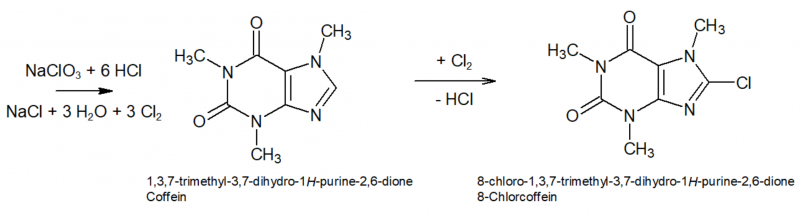
Das 8-Chlorcoffein ist hydrolyseempfindlich bzw. kann nun durch weites Chlor oxidiert und gespalten werden, sodass Dimethylalloxan entsteht. Die Spaltprodukte sind dabei Methylamin und Chlorcyan (Rochleder[7] berichtete 1849 bei seinen Versuchen bei der Umsetzung von feuchtem Coffein mit Chlorgas von einem "flüchtigen Körper, der sich durch seinen unangenehmen Geruch zu erkennen giebt. Er reizt die Augen zu Thränen und bewirkt in der kleinsten Menge einen unterträglichen Kopfschmerz in der Stirngegend"). Unter den Reaktionsbedingungen dürfte das Chlorcyan aber vermutlich sofort zu Cyansäure hydrolysiert werden die wiederum rasch zu Ammoniak und CO2 weiter zerfällt.
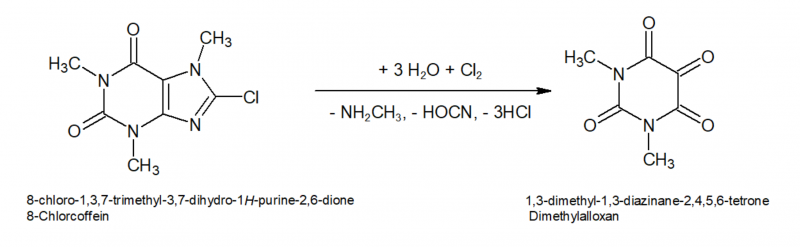
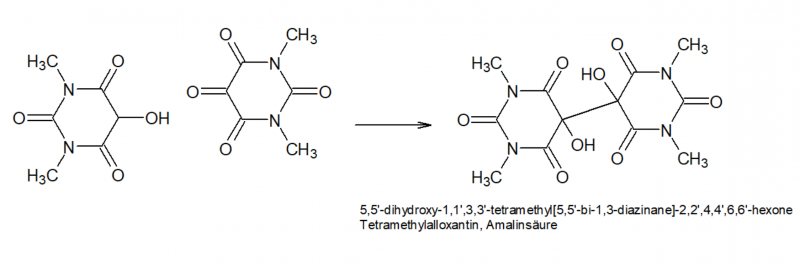
Dimethylalloxan wäre aus dem Ansatz aufgrund seiner guten Löslichkeit schwer isolierbar, daher wird es im nächsten Schritt mittels Sn(II) zu Tetramethylalloxantin reduziert. Dabei wird zunächst die Hälfte des Dimethylalloxans zu 5-Hydroxydimethylbarbitursäure (Dimethyl-Dialursäure) reduziert, diese kondensiert unter den Bedingungen mit noch freiem Dimethylalloxan zum Tetramethylalloxantin, Rochleder[7] nannte das Produkt "Amalinsäure".
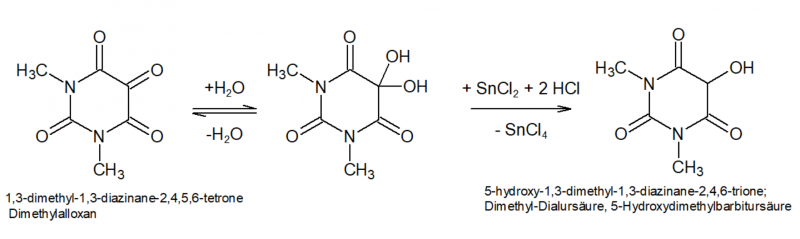
Um eine gute Ausbeute zu erzielen, darf man also keinesfalls zu viel Reduktionsmittel zusetzen, da dann weniger Produkt entsteht. Andererseits ist der oxidative Abbau des Coffeins zum Dimethylalloxan vermutlich nicht ganz stöchiometrisch bzw mit 100% Ausbeute, daher führt die Zugabe einer mehr oder weniger stöchiometrischen Menge Sn(II) zu einer zu geringen Ausbeute. Meine Vorversuche haben gezeigt, dass die nachträgliche Zugabe von H2O2 in der Kälte offensichtlich geeignet ist, hier eine gezielte "Rück-Oxidation" zu bewirken ohne wieder über das Ziel hinaus zu schießen. Erst wenn der Ansatz wieder auf Raumtemperatur ist, reagiert das H2O2 merklich mit der HCl unter erneuter Bildung von Chlor was das Produkt wieder zerstören würde.
Hier gibt es in der Literatur übrigens abweichende Darstellungen der Struktur der Amalinsäure - teils wird sie als Pinakol dargestellt (C-C-Bindung zwischen den beiden Alloxan-Ringen mit vicinalen OH-Gruppen), teils als Ketal (C-O-C-Bindung zwischen den beiden Alloxan-Ringen). Welche der beiden zutrifft bzw. ob es hier eine Tautomerie gibt dazu konnte ich keine finale Aussage finden. Davidson[13] merkte 1936 bei seinen Arbeiten zur Strukturaufklärung der Murexide jedoch an, dass wohl die Pinakol-Form vorliegen müsse, da auch O-acetylierte Alloxane die Reaktion ergeben.
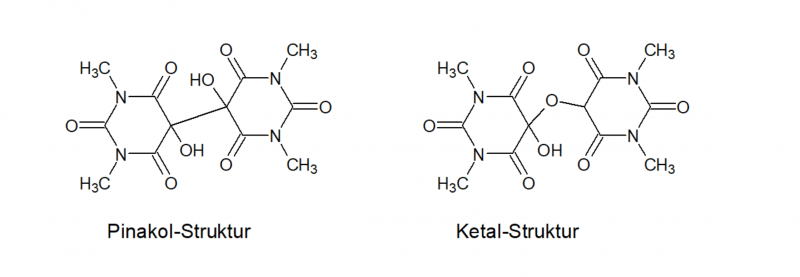
Emil Fischer[11] erklärte und systematisierte die Synthese erstmals 1882, er setzte als Reduktionsmittel H2S ein, was jedoch Schwierigkeiten mit der Entfernung des ausgeschiedenen Schwefels verursacht. Biltz[12] optimierte die Synthese 1912 indem er zur Reduktion Zinnchlorid einsetzte, er berichtete von einer Ausbeute von 84 %. Auterhoff[17] konnte 1968 im Rahmen seiner Arbeiten zur Synthese unsymmetrischer Murexide jedoch nur eine Ausbeute von 27 % bestätigen - so gesehen sind meine knapp 46 % durchaus zufriedenstellend. Ohne die Nachbehandlung mit dem H2O2 wäre die Ausbeute vermutlich ähnlich niedrig ausgefallen.
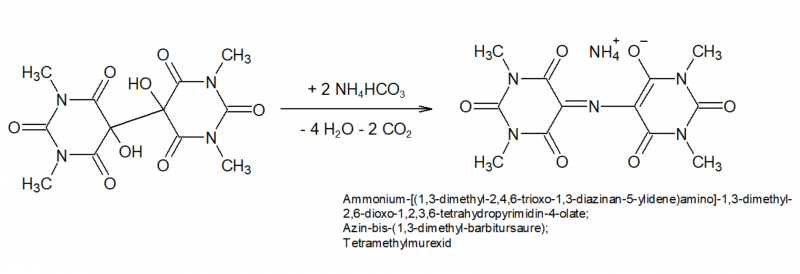
Die Umsetzung von Tetramethylalloxantin zum Murexid erfolgt sehr einfach durch direkte Umsetzung mit NH3. Auterhoff[17] leitete 8 h lang am Wasserbad trockenes NH3 auf das Alloxantin, Gysling und Schwarzenbach[16] setzten mit 20-facher Menge (40-facher molarer Überschuss) Ammoniumcarbonat um. Tatsächlich ist Ammoniumcarbonat hervorragend dafür geeignet, und die Reaktion läuft glatt ab, entstehende Feuchtigkeit hilft sicher auch bei der vollständigen "Durchtränkung" der Reaktionsmasse mit NH3. Ein Überschuss an Ammoniumcarbonat zersetzt sich in der Wärme sehr leicht und restlos in flüchtige Bestandteile, sodass eine weitere Aufreinigung nicht erforderlich ist. Eine 20-fache Menge erscheint aber auf jeden Fall hoch übertrieben.
Bilder:

Ansatz ist am Wasserbad und bereit

Ausscheidung von 8-Chlorcoffein

Durchblasen von Luft zur Vertreibung überschüssigen Chlors

Zutropfen der SnCl2-Lösung

Zwischenprodukt Tetramethylalloxantin
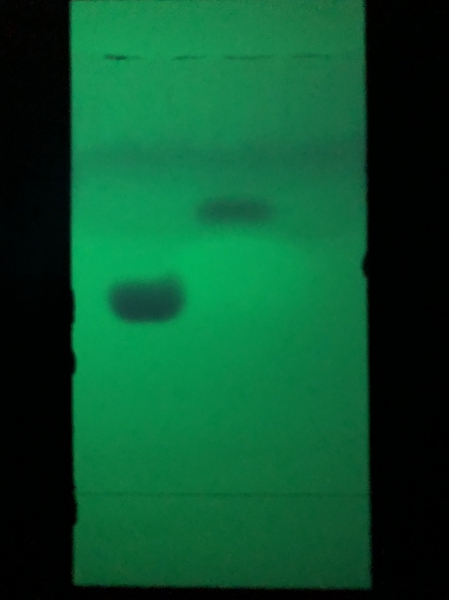
DC des Zwischenprodukts Tetramethylalloxantin (Links: Referenz Coffein, rechts: Produkt)

Umsetzung zu Tetramethylmurexid - beim Zusammengeben bildet sich sofort eine violette Färbung

Das fertige Produkt

Farbreaktionen von Tetramethylmurexid mit Metallionen: Das Reagenz, mit Cu, Ni und Co (v.l.n.r)
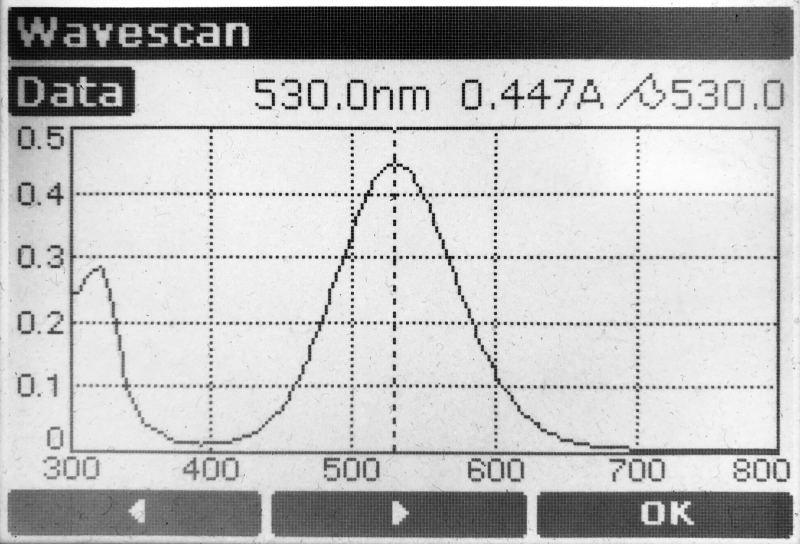
UV-VIS Spektrum des Produkts, λ-max = 530 nm

Einsatz als Indikator bei einer komplexometrischen Titration

Murexid ist bei hohem oder niedrigem pH nicht sehr beständig. Bei pH 10 zerfällt es langsam (Bilder nach 0 min, 40 min 1:40h)
Literatur:
[1] Scheele, Kongl. Vetenskaps Academiens Handlingar. (1776), 37, p.327-332
[2] Prout, Phil. Trans. (1818), p.421
[3] Wöhler, F. and Liebig, J. Ann. Pharm. (1838), 26, p.241-336
[4] Gregory, J. Prakt. Chem. (1841), 22, p.371-372
[5] Beilstein, F. Justus Liebigs Ann. Chem. (1858), 107, p.176-191
[6] Stenhouse, J., Justus Liebigs Ann. Chem.(1843), 45, p.366-372
[7] von Rochleder, F., Justus Liebigs Ann. Chem. (1849), 71, p.1-12
[8] Medicus L., Justus Liebigs Ann. Chem. (1875), 175, p.230-251
[9] Maly, R. and Hinteregger, F., Ber. Dtsch. Chem. Ges. (1881), 14, p.723-728
[10] Maly, R., Andreasch, R., Monatshefte für Chemie (1882) 3, p.92–110
[11] Fischer, E., Justus Liebigs Ann. Chem.(1882), 215, p.253-320
[12] Biltz, H., Ber. Dtsch. Chem. Ges. (1912), 45, p.3659-3675
[13] Davidson D., Epstein E., The Journal of Organic Chemistry (1936), 01(3), p.305-314
[14] Davidson D., J. Am. Chem. Soc. (1936), 58, 1821-1822
[15] Schwarzenbach, G. and Gysling, H. Helv. Chim. Acta (1949), 32, p. 1314-1325
[16] Schwarzenbach, G. and Gysling, H. Helv. Chim. Acta (1949), 32, p. 1484-1504
[17] Auterhoff, H. and Bohle, F.-J. (1968), Die Murexide der gebräuchlichen Purinderivate. Arch. Pharm. Pharm. Med. Chem., 301: 73-77