Will man nicht gerade einen gewissen Temperaturgradienten längs der Kolonne haben? Sonst macht die doch gar keinen Sinn.
Theoretisch soll eine Kolonne adiabat geführt werden, d.h. bei perfekter Isolation! Bei den ganz feinen Geräten gab es sogar einen Heizmantel, der mit feinen Reglern genau auf die Wärmeverluste eingestellt wird, sodass der Wärmeeintrag in die Kolonne sich mit den Wärmeverlusten gerade zu Null ausgleicht. So kann man bei praktisch beliebig geringen Geschwindigkeiten die volle Trennleistung einer Kolonne ausnutzen.
Etwas völlig Anderes ist die Isolation oberhalb der Kolonne. Natürlich will man oberhalb der Kolonne eine leichte Kondensation haben, um den Rücklauf zu gewährleisten, aber alles was nicht oberhalb der wirksamen Länge kondensiert, ist verschenkte Leistung, das will man verhindern. Nur wenn die gesamte Kondensation oberhalb stattfindet, stellen sich die theoretischen Gradienten ein. Alles was nahe dem Sumpfkolben kondensiert, ist verschenkt und stört nur.
Wer etwas höher siedende Verbindungen bei kleiner Belastung ohne Kolonnenkopf rektifiziert hat, weiß, dass man die nahezu optimale Kondensation meistens MIT Isolation der Brücke erreicht, sofern die Kolonne selber fast perfekt isoliert ist (beispielsweise mit verspiegeltem Vakuummantel, oder Vakuummantel + improvisierter Isolation). Dann kommt durch die improvisierte Isolation an der Brücke gerade genug Wärme, dass ein gutes Rücklaufverhältnis beibehalten werden kann. Bei Leichtsiedern lässt man das besser sein, da ist oft sogar eine kurze, nicht isolierte Kolonne oben auf die eigentliche Kolonne zu setzen um genug Rücklauf zu erreichen. Sowas ist Fummelarbeit und man muss die Ströme über die Kolonne genau im Blick haben, sonst ist die Destillation ohne Kolonnenkopf nicht reproduzierbar.
Von Vorteil ist es da immer, mit einer normalen Vigreuxkolonne zu arbeiten, das sind immer noch die robustesten Geräte, denen man am wenigsten antun kann. Vermutlich auch der Grund warum man sie am häufigsten sieht, obwohl sie eigentlich teurer sind als Füllkörperkolonnen und von allen Kolonnen mit die schlechteste Trennleistung haben.

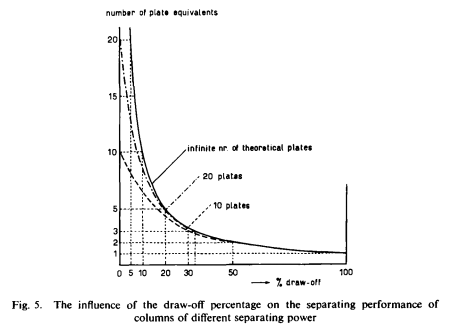

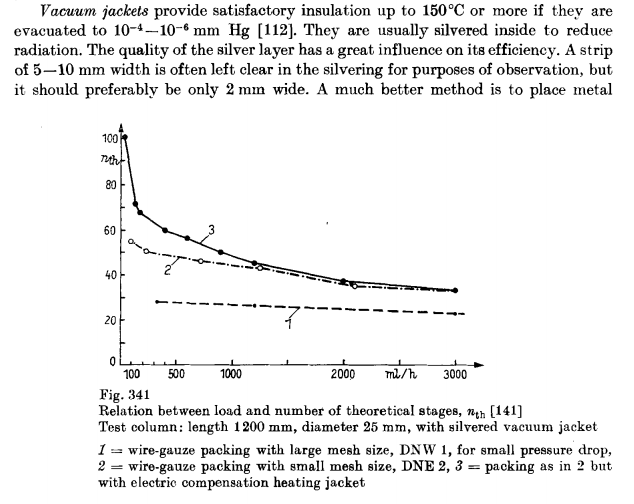
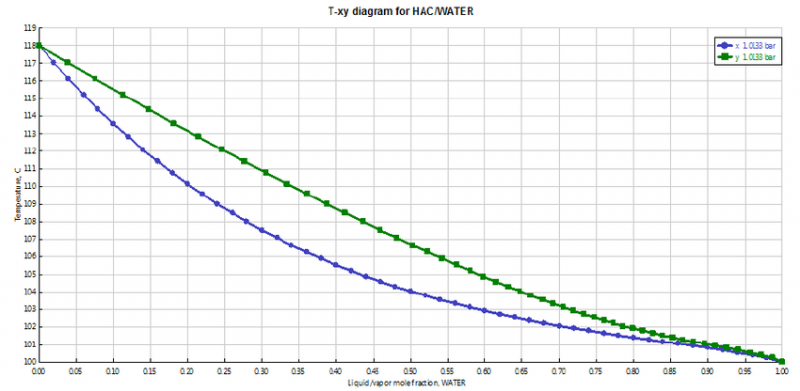
 [/img]
[/img]