Geräte:
Magnetrührer mit Heizplatte, Rührfische, 2x 400 mL Bechergläser, Apparatur zur Vakuumfiltration, Thermometer, Waage, Spatel
Chemikalien:
3-Nitrophthalsäure
Hydrazinsulfat
Natriumdithionit
Natriumhydroxid
Eisessig
Natriumacetat-Trihydrat
Glycerin
Luminol
Hinweis:
Ausführung im Abzug oder im Freien!
Durchführung:
In einem 400 mL Becherglas werden 15 g 3-Nitrophthalsäure (71 mmol, 1 eq.), 10,5 g Hydraziniumsulfat (81 mmol, 1,14 eq.), 15 g Natriumacetat-Trihydrat (110,3 mmol, 1,55 eq.) in 60 mL dest. Wasser vorgelegt. Die Suspension wird nun unter magnetischem Rühren erhitzt. Sobald sich die Lösung klärt, werden 125 mL wasserfreies Glycerin hinzugegeben. Wenn das Wasser und die entstehende Essigsäure abgedampft wurden, erhitzt man für 5 Min. auf 200-205 °C, dann kühlt man auf Raumtemperatur. Die Mischung verfärbt sich während des Erhitzens und wird dunkelrot (Abb. 1). Das 3-Nitrophthalhydrazid wird mit 240 mL Wasser gefällt, vakuumfiltriert, mit etwas dest. Wasser gewaschen und möglichst trocken gesaugt (Abb. 2).
Das noch feuchte Produkt wird in ein 400 mL Becherglas überführt, in 150 mL 10% Natronlauge gelöst (Abb. 3) und 45 g frisches Natriumdithionit (258 mmol, 3,63 eq.) hinzugefügt. Um die Reaktion zu vervollständigen wird für 5 Min. auf Kochtemperatur erhitzt (Abb. 4). Das Luminol wird nach dem Erkalten mit einem Überschuss Eisessig gefällt (pH-Wert der Lösung: 4), scharf abgesaugt, einige Male mit Wasser gewaschen und für 2 Tage zum Trocknen an der Luft ausgelegt (Abb. 5).
Ausbeute: 7,22 g (57 % d.Th.)
Analytik

Dünnschichtchromatogramm des Luminols; Laufmittel: Dichlormethan/Methanol/25% Ammoniakwasser (9ml/2ml/5 Tropfen); stationäre Phase: Kieselgel 60 auf Aluminium mit F254 Fluoreszenzindikator; Detektion: 254 nm UV-Licht

Selbe DC-Platte bei 366 nm UV-Licht
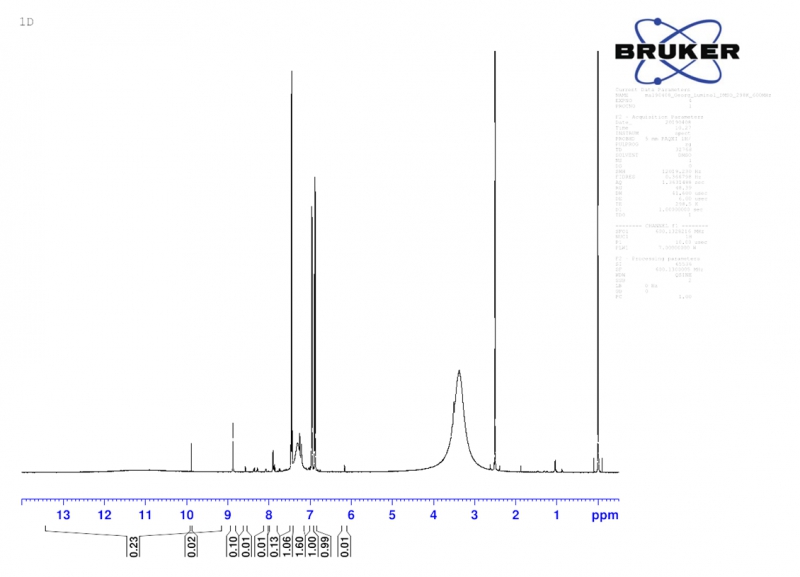
H-NMR-Spektrum des Luminols in DMSO-d6, aufgenommen mit einem Bruker 600 MHz NMR-Spektrometer
Entsorgung:
Alle Lösungen kommen in den Behälter für wässrige, organische, halogenfreie Abfälle.
Erklärung:
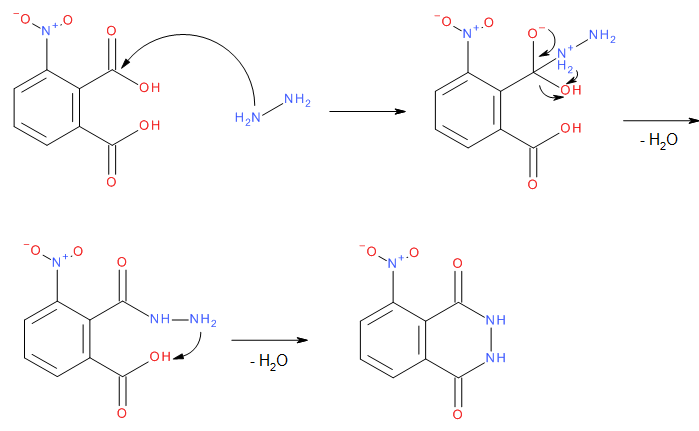
Hydrazinsulfat reagiert mit dem Natriumacetat zu Hydrazinacetat und Natriumsulfat. Ersteres zersetzt sich bei höherer Temperatur und es bilden sich Essigsäure und Hydrazin.
Das in Situ gebildete Hydrazin greift dann die Carbonylgruppe der 3-Nitrophthalsäure an. Die OH-Gruppe wird protoniert und als Wasser abgespalten. Das zweite Stickstoff Atom greift dann die zweite Carbonylgruppe der 3-Nitrophthalsäure an und es wird ein weiteres Molekül Wasser abgespalten, wodurch die Zielverbindung 3-Nitrophthalhydrazid entsteht.
Die Nitrogruppe wird dann mit Natriumdithionit reduziert, 3-Aminophthalhydrazid (Luminol) wird gebildet.
Bilder:

Erhitzen auf 200 °C

Abfiltrieren des 3-Nitrophthalhydrazids

Lösen des 3-Nitrophthalhydrazids in 10 % Natronlauge

Erhitzen auf Kochtemperatur, um die Reduktion zu vervollständigen

Abfiltrieren des Luminols





{kind=link}