Hm, es wurde ja auch von Problemen bei diesem Schritt geschrieben, aber eher in Richtung schlechte Ausbeute und viele Nebenreaktionen...
Bist du genau nach den Angaben in der experimental section vorgegangen? Die Wasserfreiheit des Eduktes und des Kupfer(I)iodids könnten entscheidend sein... (Evtl. auch das 2,4,6-Trimethylpyridin vorher trocknen)
Retrosynthese
Moderator: Moderatoren

Ja, war zu erwarten.
Naja nicht ganz, habe die Menge an Collidin etwas erniedrigt. Verstehe auch nicht, warum man solche gigantischen Mengen brauchen soll. Soviel ist ja in unserem ganzen Arbeitskreis gar nicht vorhanden, warscheinlich... Naja wir werden sehen.
Übrigens: Weiß jemand, wo man auf die Schnelle eine kleine Menge Ethen herbekommt? Im Baumarkt bekommt man ja viele Gase wie Argon etc. in so kleinen 2L Flaschen? Gibts auch Ethen?
Naja nicht ganz, habe die Menge an Collidin etwas erniedrigt. Verstehe auch nicht, warum man solche gigantischen Mengen brauchen soll. Soviel ist ja in unserem ganzen Arbeitskreis gar nicht vorhanden, warscheinlich... Naja wir werden sehen.
Übrigens: Weiß jemand, wo man auf die Schnelle eine kleine Menge Ethen herbekommt? Im Baumarkt bekommt man ja viele Gase wie Argon etc. in so kleinen 2L Flaschen? Gibts auch Ethen?
-
Calciumcitrat
- Illumina-Mitglied
- Beiträge: 170
- Registriert: Montag 11. April 2011, 16:57
-
Cumarinderivat

- Illumina-Mitglied
- Beiträge: 625
- Registriert: Samstag 2. Januar 2016, 16:47
-
Glaskocher
- Illumina-Mitglied
- Beiträge: 2534
- Registriert: Dienstag 27. Oktober 2015, 22:17
- Wohnort: Leverkusen
Ethen ist tatsächlich etwas kniffelig im Kleinlabor. Man bekommt es als technisches Gas in 10L-Stahlflaschen, teuren Lecturebottles oder im "Eigenbau".
Für letzteres tropft man Ethanol in überschüssige, auf 170°C erhitzte konz. Schwefelsäure.
Siehe LINKs
Eventuell muß man das Gas noch etwas reinigen, wenn man es chemisch nutzen will. Test beweist...
Für letzteres tropft man Ethanol in überschüssige, auf 170°C erhitzte konz. Schwefelsäure.
Siehe LINKs
Eventuell muß man das Gas noch etwas reinigen, wenn man es chemisch nutzen will. Test beweist...
Die Methoxylierung funktioniert nun einwandfrei, seit ich meine Natriummethoxid-Lösung selbst aus Natrium und wasserfreiem MeOH hergestellt habe (2,7M), statt diese alte 0,5M Brühe zu verwenden (hatte sogar AcrosSeal Verschluss  ). Außerdem jetzt die volle Menge an Collidin verwendet. Die Reagenzien wurden nicht speziell getrocknet und die Reaktion auch nicht unter Schutzgas durchgeführt. Wenn man anfängt rückflusszukochen, ändert sich die Farbe der Lösung innerhalb der ersten 10 Minuten recht schnell von grünlich nach kupferbraun. Das scheint ein Indiz zu sein, dass die Reaktion funktioniert, da dies beim ersten mal nicht der Fall war.
). Außerdem jetzt die volle Menge an Collidin verwendet. Die Reagenzien wurden nicht speziell getrocknet und die Reaktion auch nicht unter Schutzgas durchgeführt. Wenn man anfängt rückflusszukochen, ändert sich die Farbe der Lösung innerhalb der ersten 10 Minuten recht schnell von grünlich nach kupferbraun. Das scheint ein Indiz zu sein, dass die Reaktion funktioniert, da dies beim ersten mal nicht der Fall war.
Die Aufreinigung durch Säulen war zum Glück sehr leicht, da die Spots schon auf der TLC weit auseinander lagen. Hab verschiedene Eluenten getestet und Dichlormethan/Hexan 1:1 als am besten befunden. In der Vorschrift wird mit Toluol gearbeitet- aber ähh nein! Ausbeute lag nach dem Säulen bei 60%.
Ausbeute lag nach dem Säulen bei 60%.
Bin jetzt mal seeehr gespannt auf den letzten Schritt! Ich schätze, dass das aktive Agens dort "naszierender" Wasserstoff ist? Eine richtige Birch-Reduktion ist es ja nicht, da man nicht in flüssigem Ammoniak arbeitet, sondern in warmen Ethanol. Oder findet die Reaktion doch an der Oberfläche des Natriums statt, während es sich auflöst?
Es muss doch kleinere Ethen-Kartuschen geben? Wobei, eventuell brauche ich es eh nicht mehr...
Bei dem anderen Syntheseweg zum 5,6-Dimethoxy-2-tetralon stecke ich bei dem beta-Keto-Sulfoxid fest, welche ich versucht hab zu säulen, was vielleicht keine Idee war. Kann die Polarität des Moleküls schlecht einschätzen. Naja mal schaun. Da der Naphtholweg sehr gut funktioniert, werd ich warscheinlich da nicht weitermachen...
Die Aufreinigung durch Säulen war zum Glück sehr leicht, da die Spots schon auf der TLC weit auseinander lagen. Hab verschiedene Eluenten getestet und Dichlormethan/Hexan 1:1 als am besten befunden. In der Vorschrift wird mit Toluol gearbeitet- aber ähh nein!
Bin jetzt mal seeehr gespannt auf den letzten Schritt!
Da kommt bei mir aber keine Begeisterung auf!Für letzteres tropft man Ethanol in überschüssige, auf 170°C erhitzte konz. Schwefelsäure.
Bei dem anderen Syntheseweg zum 5,6-Dimethoxy-2-tetralon stecke ich bei dem beta-Keto-Sulfoxid fest, welche ich versucht hab zu säulen, was vielleicht keine Idee war. Kann die Polarität des Moleküls schlecht einschätzen. Naja mal schaun. Da der Naphtholweg sehr gut funktioniert, werd ich warscheinlich da nicht weitermachen...
-
Calciumcitrat
- Illumina-Mitglied
- Beiträge: 170
- Registriert: Montag 11. April 2011, 16:57
Das freut mich sehr!
Die "Birch"-Reduktion wird erstmalig hier so beschrieben: http://pubs.rsc.org/en/content/articlep ... age=search
Also schon etwas ältere Chemie. Genau wie das Konzept von nasc. Wasserstoff. Ich halte davon wenig. Das wird hier wohl irgendwas in Richtung Einzel-Elektronen-Reduktion... siehe auch: http://www.sciencedirect.com/science/ar ... via%3Dihub
Die Trennung des Produktes vom auch entstehenden 6-Methoxy-2-tetralon wird als schwierig beschrieben, vielleicht könnte das auf präp. HPLC hinauslaufen, ich würde es aber zuerst über eine Säule versuchen.
Die "Birch"-Reduktion wird erstmalig hier so beschrieben: http://pubs.rsc.org/en/content/articlep ... age=search
Also schon etwas ältere Chemie. Genau wie das Konzept von nasc. Wasserstoff. Ich halte davon wenig. Das wird hier wohl irgendwas in Richtung Einzel-Elektronen-Reduktion... siehe auch: http://www.sciencedirect.com/science/ar ... via%3Dihub
Die Trennung des Produktes vom auch entstehenden 6-Methoxy-2-tetralon wird als schwierig beschrieben, vielleicht könnte das auf präp. HPLC hinauslaufen, ich würde es aber zuerst über eine Säule versuchen.
Die Reduktion mit Natrium funktioniert super! Leider bildet sich nur zu ca. 2/3 das gewünschte Keton und zu 1/3 auch das 6-Methoxy-2-tetralon, wie ja auch in der Vorschrift schon angegeben war. Ich habe versucht zu säulen, habe aber keine Trennung hinbekommen, obwohl die beiden Ketone im LC-MS Spektrum durchaus einen gewissen Unterschied in den Elutionszeiten zeigen. Um zu meinen nächsten Zielmolekülen zu gelangen muss ich das Keton reduktiv aminieren (mit einem Phenalkylamin). Dies habe ich mit dem verunreinigten Keton getan und natürlich wie erwartet, die beiden sekundären N-Phenalkylamine erhalten.
Diese ließen sich auch nicht per Säule trennen (war sogar noch schlimmer, verschwimmen in der LC-MS zu einem Peak). Da ich das sekundäre N-Phenalkylamin in seine Enantiomere trennen muss und dafür die chirale präparative HPLC brauche, überlege ich, das Gemisch direkt dort einzusetzen. Nach den ersten Versuchen (analytische chirale HPLC) hat sich jetzt gezeigt, dass sich die beiden Amine trennen und auch jeweils noch in ihre Enantiomere. Ich sehe also 2 Peakpaare, die jeweils die selbe Signalintensität zeigen (50:50 = Racemat). Verunreinigungen sind natürlich auch noch dabei, aber zum Glück nicht in dem Bereich, wo die Produkte eluieren!
Ich halte das momentan für die beste Option, wenn ich nämlich das Keton aufreinigen wollte, müsste ich dies per "normaler" präp. HPLC tun und nach der reduktiven Aminierung nochmal eine chirale präp. HPLC. Es wäre natürlich super, wenn sich dies auf einen Schritt verkürzen ließe.
Diese ließen sich auch nicht per Säule trennen (war sogar noch schlimmer, verschwimmen in der LC-MS zu einem Peak). Da ich das sekundäre N-Phenalkylamin in seine Enantiomere trennen muss und dafür die chirale präparative HPLC brauche, überlege ich, das Gemisch direkt dort einzusetzen. Nach den ersten Versuchen (analytische chirale HPLC) hat sich jetzt gezeigt, dass sich die beiden Amine trennen und auch jeweils noch in ihre Enantiomere. Ich sehe also 2 Peakpaare, die jeweils die selbe Signalintensität zeigen (50:50 = Racemat). Verunreinigungen sind natürlich auch noch dabei, aber zum Glück nicht in dem Bereich, wo die Produkte eluieren!
Ich halte das momentan für die beste Option, wenn ich nämlich das Keton aufreinigen wollte, müsste ich dies per "normaler" präp. HPLC tun und nach der reduktiven Aminierung nochmal eine chirale präp. HPLC. Es wäre natürlich super, wenn sich dies auf einen Schritt verkürzen ließe.
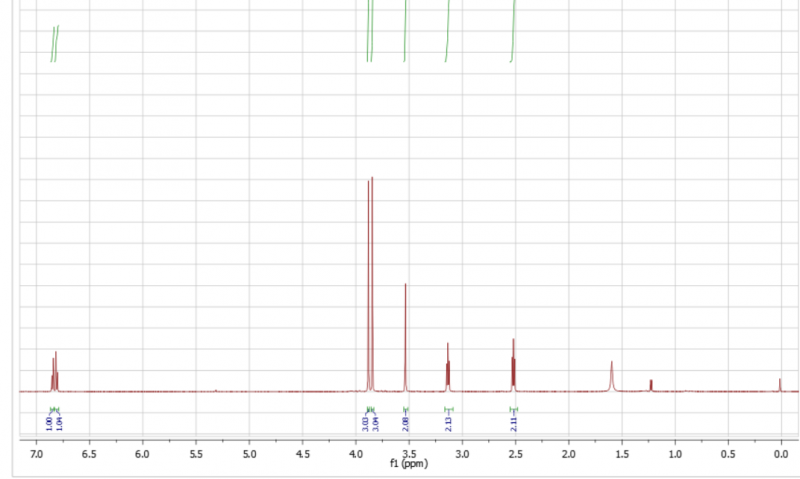
Weiß jemand, ob man Bisulfit-Addukte direkt reduktiv aminieren kann, indem man sie in einem polaren aprotischen LM löst (Dioxan oder so) und dann direkt ein Äquivalent Triethylamin UND ein Äquivalent umzusetzendes Amin zugibt? Und natürlich noch Natriumtriacetoxyborhydrid oder Natriumcyanoborhydrid. Das freie Keton ist offensichtlich instabil und ich will es direkt abfangen.
-
Calciumcitrat
- Illumina-Mitglied
- Beiträge: 170
- Registriert: Montag 11. April 2011, 16:57
Das wird in der Literatur als instabil beschrieben, deswegen konnte ich es von einer Firma auch nur als Bisulfit kaufen. Was da wirklich dran ist- keine Ahnung. Ich hab das Keton wegen dieser Information immer recht schnell verarbeitet.
Übrigens hab ich ausgerechnet, dass ich für die Synthese von 2g des Ketons (das 6,7-Dimethoxy-2-tetralon, also nicht das, dass mit dem Naphtholweg hergestellt wurde) 200 mL Ethen brauche. Das muss doch mit Ethanol/Schwefelsäure locker hinzubekommen sein? Würde direkt nach der Mischung eine Gaswaschflasche schalten, und das Gas dann per Ballon auffangen und direkt an die Reaktionsmischung hängen. Klingt doch realistisch oder?
Übrigens hab ich ausgerechnet, dass ich für die Synthese von 2g des Ketons (das 6,7-Dimethoxy-2-tetralon, also nicht das, dass mit dem Naphtholweg hergestellt wurde) 200 mL Ethen brauche. Das muss doch mit Ethanol/Schwefelsäure locker hinzubekommen sein? Würde direkt nach der Mischung eine Gaswaschflasche schalten, und das Gas dann per Ballon auffangen und direkt an die Reaktionsmischung hängen. Klingt doch realistisch oder?

Jo, finde auch dass das realistisch klingt. Bei der Schwefelsäure-Prozedur können allerdings auch SO2 und CO2 entstehen (dann sollte in die Waschflasche Natronlauge). Mit Phosphorsäure soll es bequemer gehen. Da entsteht als Nebenprodukt nur etwas Diethylether, den man leicht entfernen kann. Noch ne Möglichkeit wäre die katalytische Zersetzung über Aluminiumoxid, aber da sind die Verunreinigungen vielleicht größer.