Ester von Phenolen oder tertiären Alkoholen lassen sich aufgrund mangelnder Carbonylaktivität nicht durch direkte Alkoholyse der Carbonsäure herstellen. Man greift deshalb auf acidere Edukte (stärker positivierter Carbonylkohlenstoff durch -I-Effekt), wie Carbonsäureanhydride oder Carbonsäurechloride zurück.
Geräte:
Rückflusskühler, 250 ml Rundkolben, Trockenrohr, Magnetrührer, Magnetrührstab, Wasserbad, Becherglas
Chemikalien:
Essigsäureanhydrid
Phenol
Schwefelsäure
Natriumcarbonat
Natriumsulfat
Essigsäurephenylester

Durchführung:
15,7 mL Essigsäureanhydrid werden in einer Rückflussapparatur zusammen mit 13,5 g Phenol versetzt. Als Katalysator gibt man 5 Tropfen konz. Schwefelsäure hinzu. Man stellt das Wasserbad an und lässt das Reaktionsgemisch in siedendem Wasser 2 Stunden lang reagieren. Das Phenol löst sich bei Erwärmung sofort vollständig in dem Essigsäureanhydrid.
Anfangs ist die Lösung etwas rosa gefärbt, was sich im weiteren Reaktionsverlauf abschwächt, sodass schließlich die homogene Lösung farblos erscheint. Ist die Reaktion beendet, gießt man das Rohprodukt in Eiswasser und lässt solange stehen bis sich zwei Phasen gebildet haben.
Die organische Phase wird abgetrennt und im Scheidetrichter mit Natriumcarbonatlösung gewaschen. Das entstehende Kohlenstoffdioxid lässt man ab, indem man den Scheidetrichter umdreht und den Hahn öffnet. Dieser Vorgang wird so oft wiederholt, bis kein Gas mehr aus dem Hahn entweicht. Danach trennt man die organische Phase im Scheidetrichter ab, wäscht sie anschließend noch zweimal mit destillierten Wasser und trocknet sie dann über Natriumsulfat.
Zur weiteren Aufarbeitung bietet sich eine Vakuumdestillation an, welche mir aber leider nicht möglich ist.
Ausbeute: 14,1 g (72,2% d.Th.) (Literaturausbeute: 75% (Organikum))
Entsorgung:
Der Essigsäurephenylester kommt zu den halogenfreien Lösungsmittelabfällen.
Erklärung:
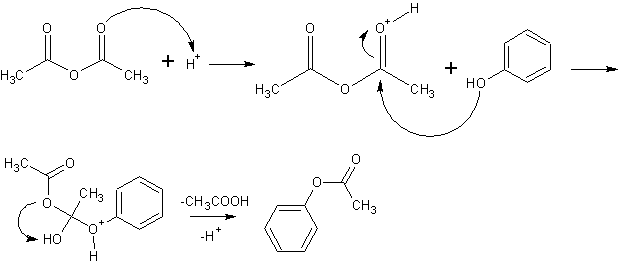
Im ersten Schritt wird der Carbonylsauerstoff protoniert (Protonen von der Schwefelsäure). Durch die Protonierung wird eine verstärkte positive Ladung am Carbonylkohlenstoffatom erreicht. Im zweiten Schritt klappen zwei Bindungselektronen nach oben, sodass der Hydroxyl-Sauerstoff des Phenols nucleophil angreifen kann. Die neue Esterbindung C-O ist geknüpft. Das entstandene Oxoniumion in Schritt 3 wird durch Deprotonierung beseitigt (Katalysatorrückbildung). Des weiteren wird Essigsäure aus dem Hydroxylwasserstoff und dem Acetation abgespalten und so in Schritt 4 der Ester freigesetzt.
Bilder:

Die Reaktionsapparatur

Das Reaktionsgemisch nach einer Stunde beim Rückflusskochen

Das Rohprodukt im Kolben

Der abgefüllte Essigsäurephenylester



